


Abstract
Background: Transforming growth factor β (TGF-β) plays a complex role in breast carcinogenesis. Initially functioning as a tumor suppressor, this cytokine later contributes to the progression of malignant cells by enhancing their invasive and metastatic potential as well as suppressing antitumor immunity. The purpose of this study was to investigate the efficacy of SM16, a novel small molecule ALK5 kinase inhibitor, to treat a highly metastatic, TGF-β-producing murine mammary carcinoma (4T1). Materials and Methods: Mice bearing established 4T1 tumors were treated with SM16 intraperitoneally (i.p.) or orally, and primary and metastatic tumor growth was assessed. Results: SM16 inhibited Smad2 phosphorylation in cultured 4T1 tumor cells as well as primary and metastatic 4T1 tumor tissue. Blockade of TGF-β signal transduction in 4T1 tumor cells by SM16 prevented TGF-β-induced morphological changes and inhibited TGF-β-induced invasion in vitro. When delivered via daily i.p. injection or orally through mouse chow, SM16 inhibited the growth of primary and metastatic 4T1 tumors. Splenocytes isolated from mice on the SM16 diet displayed enhanced IFN-γ production and antitumor CTL activity. Furthermore, SM16 failed to inhibit the growth and metastasis of established 4T1 tumors in immunodeficient SCID mice. Conclusion: Taken together, the data indicate that the antitumor efficacy of SM16 is dependent on an immune-mediated mechanism and that SM16 may represent a safe and effective treatment for metastatic breast cancer.
Transforming growth factor-β is a multifunctional cytokine belonging to the TGF-β superfamily of secreted cytokines that plays a complex role in breast carcinogenesis (1-4). Initially TGF-β acts as a tumor suppressor by inhibiting cell proliferation, but as tumor progression occurs, malignant cells become resistant to its growth inhibitory effects and begin to overexpress and secrete large amounts of this cytokine (2, 5, 6). Breast tumors, like many other epithelial tumors have been shown to produce elevated levels of TGF-β that are associated with disseminated disease, tumor recurrence and poor clinical outcome (7-14).
The role of TGF-β in mammary tumor progression has been well documented and includes promoting tumor cell motility by inducing the epithelial to mesenchymal transition (EMT), stimulation of angiogenesis, promotion of extracellular matrix degradation through the induction of matrix metalloprotease (MMP) expression, and inhibition of antitumor immunity (14-17). Our laboratory showed that tumor-derived TGF-β directly contributes to progression of murine 4T1 mammary tumors by enhancing their invasive and metastatic potential (16). Expression of a dominant negative TGF-β receptor type II significantly reduced the ability of 4T1 tumor cells to establish spontaneous pulmonary metastases (16). In addition, we showed that TGF-β interferes with the induction of effective antitumor immune responses by impairing the immunostimulatory capacity of dendritic cells (17). In attempting to override the deleterious effects of tumor-derived TGF-β, our laboratory demonstrated that neutralization of TGF-β with an anti-sense TGF-β transgene in combination with the TGF-β-neutralizing monoclonal antibody 2G7 enhanced the efficacy of tumor lysate-pulsed DC to inhibit the growth of established 4T1 tumors (17).
The ability of tumor-derived TGF-β to promote tumor progression directly by increasing the invasive and metastatic potential of tumor cells and indirectly by inhibiting antitumor immunity points to the TGF-β signaling pathway as an important therapeutic target in treating breast cancer as well as other malignancies. As a result, a number of small molecule TGF-β receptor type I (ALK5) kinase antagonists have recently been developed and have shown promise in early pre-clinical studies (18-24). These compounds target the TGF-β signaling pathway by binding to the ATP-binding pocket of ALK5 thereby preventing TGF-β-mediated downstream signaling events. Furthermore, some of these inhibitors have shown antitumor efficacy in murine models of glioma, mesothelioma, head and neck cancer, and breast cancer (20-24).
SM16 is a novel, orally bioavailable kinase inhibitor that binds to the ATP-binding pocket of ALK5, inhibiting its activation (22-24). When tested against a panel of 35 unrelated kinases, SM16 was shown to be highly selective for ALK5 (IC50=64 nM) and only moderately inhibited the activity of p38α and Raf.
In this study we investigated the use of SM16 to treat established primary and metastatic 4T1 mammary tumors.
Materials and Methods
Cell lines. The 4T1 tumor cell line is a poorly immunogenic, highly metastatic variant of 410.4, a tumor subline isolated from a spontaneous mammary tumor that developed in a BALB/cfC3H mouse (25). It was kindly provided by Dr. Fred Miller of the Michigan Cancer Foundation (Detroit, MI, USA). The murine leukemia cell line 12B1 was purchased from ATCC (Rockville, MD, USA). The cells were maintained for a limited time in vitro by passage in Iscove's modified Dulbecco's medium (IMDM) (JRH Biosciences, Lenexa, KS, USA) containing penicillin (100 U/ml), streptomycin (100 μg/ml), fungizone (0.75 μg/ml) and 10% fetal bovine serum (FBS) (Gemini Bio-Products, Woodland, CA, USA). The small molecule ALK5 kinase inhibitor, SM16, was kindly provided by Biogen Idec (Cambridge, MA, USA).
Animals. Six- to 8-week-old female BALB/c mice or C.B17/IcrACCscid mice were purchased from the National Cancer Institute, Frederick Cancer Research Facility (Frederick, MD, USA) and the Experimental Mouse Shared Service at the University of Arizona (Tucson, AZ, USA) respectively. All mice were housed at the University of Arizona Animal Care Facilities in accordance with the Principles of Animal Care (NIH publication no. 85-23, revised 1985). The Animal Use Committees of the University of Arizona approved all protocols in compliance with the Guide for the Care and Use of Laboratory Animals. Mice were fed food and water ad libitum.
Preparation of lysates for Western immunoblotting. To obtain cell lysates, 4T1 cells grown overnight in IMDM containing 0.5% FBS were treated with SM16 at a final concentration of 0.5, 2.5, or 5 μM for 30 min followed by exposure to 2 ng/ml rhTGF-β1 (Peprotech, Rocky Hill, NJ, USA) for 45 min. The cells were washed twice with ice cold PBS, scraped into RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.25% Na-deoxycholate, 1 mM EDTA) containing protease inhibitors (1 mM phenylmethylsulphonyl fluoride (PMSF), 1 mM NaF, 1 mM Na3VO4, 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 μg/ml pepstatin) and Phosphatase Inhibitor Cocktail 1 (Sigma, St. Louis, MO, USA), and forced through a 21-gauge needle. The lysates were then incubated on ice for 10 min before clarification by centrifugation at 14,000 × g for 15 min at 4°C. Protein in the supernatant was quantified using the BCA Protein Assay Kit (Pierce, Rockford, IL, USA).
To obtain tumor tissue lysates, tumors were generated by orthotopic injection of 5×104 4T1 cells into the mammary fat pad of BALB/c mice. Primary tumors were allowed to reach an average size of ~150 mm2 before the mice were either injected intraperitoneally (i.p.) with a single dose of 10 or 40 mg/kg of SM16 in Captisol (CyDex, INC., Lenexa, KS, USA) or put on a special diet containing SM16 (0.45 g SM16/kg food) (Research Diets, New Brunswick, NJ, USA). One hour after SM16 injection or 24 hours after transfer to SM16 diet, primary tumors or lungs were excised, washed in ice-cold PBS, minced on ice and homogenized in CelLytic MT mammalian tissue lysis reagent (Sigma) containing protease inhibitors and Phosphatase Inhibitor Cocktail 1 (Sigma) using Dounce tissue grinders. The resulting lysate was forced through a 21-gauge needle, incubated on ice for 10 min, and centrifuged twice at 14,000 × g for 15 min at 4°C. Protein in the supernatant was quantified using the BCA Protein Assay (Pierce).
Western immunoblotting. Proteins (35 μg) from the lysates were resolved by 10% SDS PAGE and electro-transferred to polyvinylidene fluoride (PVDF) membrane. Nonspecific binding sites were blocked in TBST/Milk (Tris-buffered saline containing 0.1% Tween-20 and 5% nonfat powdered milk). The membranes were immunoblotted with rabbit anti-phosphorylated Smad2 polyclonal antibody (1:500 in TBST/Milk) (US Biological, Swampscott, MA, USA) and membrane-bound pSmad2 was visualized with goat anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody (1:500 in TBST/Milk) (Pierce) using the Supersignal West Pico Chemiluminescent Substrate (Pierce). The membranes were subsequently stripped using Restore Western Blot Stripping buffer (Pierce) according to the manufacturer's instructions and reprobed with a mouse anti-Smad2/3 monoclonal antibody (1:500 in TBST/Milk) (BD PharMingen, San Diego, CA, USA). Membrane-bound total Smad2/3 was visualized with a goat anti-mouse-HRP-conjugated secondary antibody (1:500 in TBST/MLK) (Upstate Biotechnology, Lake Placid, NY, USA) and Supersignal (Pierce).
Immunofluorescence microscopy. 4T1 cells (1×105) were grown on 0.17 mm-thick glass coverslips for 24 h in complete IMDM containing 10% FBS. Cells were treated with 5 μM SM16 for 30 min in complete IMDM containing 0.5% FBS followed by exposure to 2 ng/ml rhTGF-β1 for 24 h. Cells were fixed in 3.7% methanol-free formaldehyde for 10 min at room temperature and permeabilized with 0.1% Triton® X-100 in PBS for 5 min. For visualization of F-actin, fixed cells were stained with 200 μl of a 1:40 dilution of FITC-phalloidin (Invitrogen, Eugene, OR, USA) in PBS containing 1% bovine serum albumin (BSA) for 20 min at room temperature. All samples were mounted using Aqua-Poly/Mount mounting media (Polysceinces, Warrington, PA, USA). Slides were visualized and photographed using a Nikon Eclipse TE 2000-U inverted fluorescence microscope (Nikon Instruments Inc., Melville, NY, USA) at ×1,000 magnification.

Inhibition of Smad2 phosphorylation by SM16 in cultured 4T1 tumor cells and 4T1 tumors. A, 4T1 cells were treated with SM16. Lysates were prepared and pSmad2 was detected by Western blot analysis. B, Mice bearing established 4T1 tumors were injected i.p. with a single dose of SM16 (10 or 40 mg/kg) or vehicle (Captisol). One hour after treatment, primary tumors or lungs were homogenized and pSmad2 was detected by Western blot analysis.
In vitro invasion assay. BD Biocoat Growth Factor Reduced Matrigel Invasion Chambers (24-well insert; 8 μm pore size; BD Biosciences, Bedford MA, USA) were rehydrated by adding 0.5 ml of complete IMDM to the upper chamber of the insert for 2 h at 37°C. 4T1 cells were pre-treated with 5 μM SM16 in IMDM containing 0.5% FBS for 30 min. Subsequently, the cells were seeded at a density of 1×105 cells/well in triplicate in the upper chamber of the insert to which 2 ng/ml rhTGF-β1 was added. Twenty-four hours later, the cells in suspension were aspirated, the transwells were washed twice, and the cells that were adherent to the top of the inserts were removed by scraping the upper surface with a cotton-tipped applicator. Cells that had migrated through the transwell onto the lower surface were fixed and stained using the DiffQuick staining kit (Dade Behring, Newark, DE, USA) according to the manufacturer's protocol. Cells were counted visually in nine random fields of view under a microscope at ×200 magnification.
Animal studies. For the i.p. treatment studies, 6- to 8-week-old female BALB/c mice (N=10 per group) were injected subcutaneously (s.c.) with 5×104 4T1 cells in 100 μl PBS into the mammary fat pad on day 0. When tumors had reached an average size of ~9-10 mm2, the mice were injected i.p. with SM16 at a concentration of 40 mg/kg body weight in 200 μl of 20% Captisol (CyDex Inc). SM16 injections were continued at the same dose daily until day 28 when the study was terminated. Control animals received daily injections of 20% Captisol (vehicle). For the oral treatment studies, 6-week-old female BALB/c mice (N=10 per group) or immunodeficient C.B17/IcrACCscid mice (N=5 per group) bearing ~9-10 mm2 tumors were fed either normal chow or chow containing SM16 at a concentration of 0.45 g SM16/kg food (Research Diets). In all cases, tumor growth was monitored three times weekly by measuring tumor length (L) and width (W) and tumor size (mm2) was calculated using the formula L × W. Mice and chow were weighed every 4 days to monitor toxicity and food consumption. Pulmonary metastatic nodules were enumerated in mice at the time of sacrifice by staining lungs with India ink and Fekete's solution (26).
Analysis of serum SM16 levels. Serum was isolated from mice in each treatment group in the oral SM16 studies at various time points by terminal cardiac puncture. The concentration of SM16 in plasma was analyzed by high-performance liquid chromatography on a Zorbax SB-C8 3.5-Am (2.1×50 mm) column (Agilent, Palo Alto, CA, USA) followed by mass spectrometry (MS) on a triple quadrupole mass spectrometer (SCIEX API 4000; Applied Biosystems, Foster City, CA, USA) equipped with a Turbo Ion Spray probe operated in positive ion mode. Plasma SM16 was first subjected to solid-phase extraction on an Oasis HLB AElution SPE plate (Waters, Milford, MA, USA) preconditioned with methanol and water (SPE), washed with 5% methanol, and eluted with acetonitrile/isopropanol (40:60). Samples were further diluted with 50:50:0.1 acetonitrile/water/formic acid (v/v/v) before analysis by liquid chromatography-MS/MS using multiple reaction monitoring. Data were collected and processed using Analyst version 1.4.1 (Applied Biosystems).

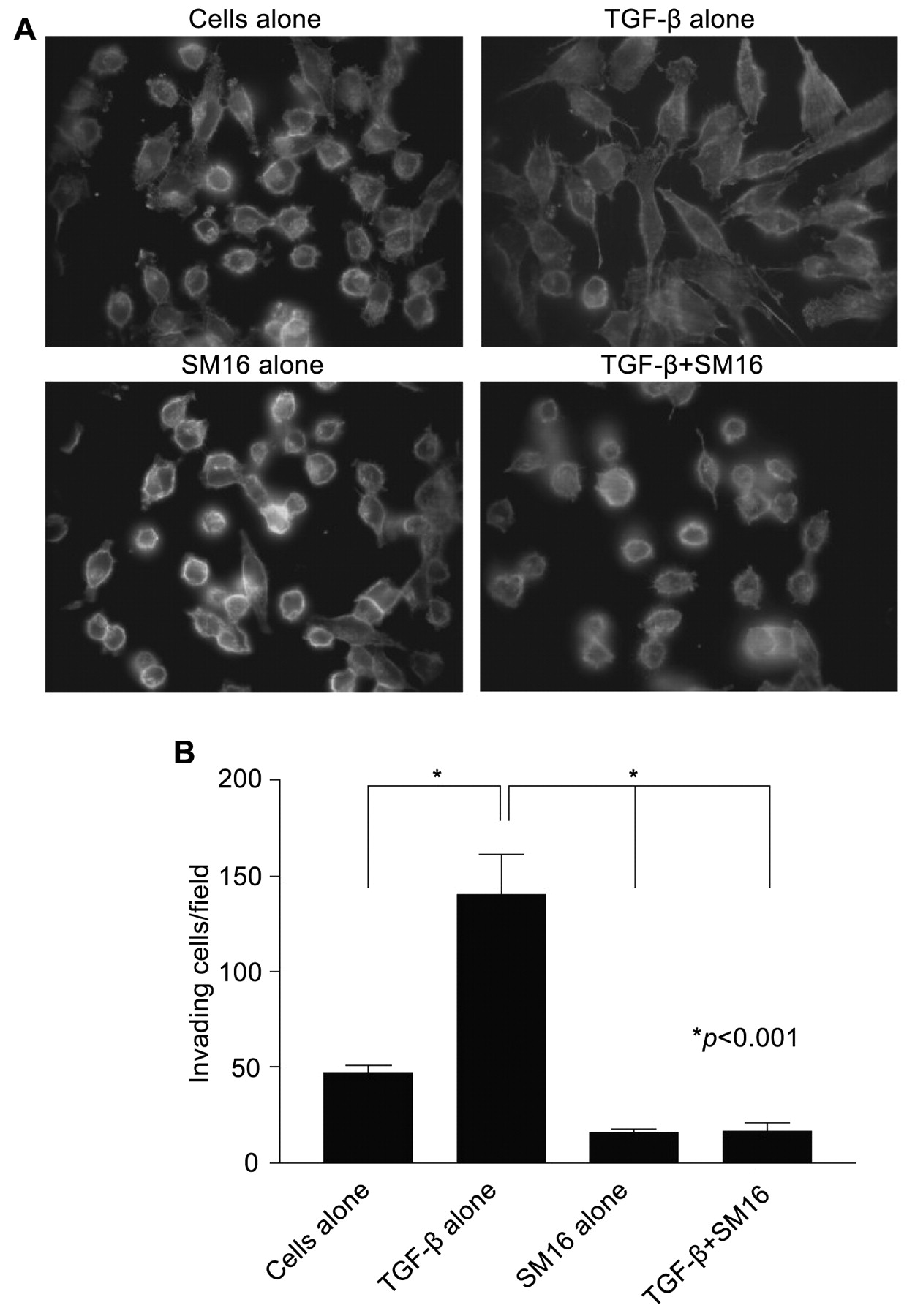
Effect of SM16 on TGF-β-induced morphological changes and invasion in vitro. A, 4T1 cells were treated with rhTGF-β1 in the presence or absence of SM16 for 24 h. Cells were then stained with FITC-conjugated phalloidin and visualized on a Nikon Eclipse TE 2000-U microscope at ×1,000 magnification. B, 4T1 cells were plated in growth factor-reduced Matrigel invasion cell culture inserts for 24 h in the presence of rhTGF-β1 with or without SM16. Invading cells were stained with the DiffQuick staining kit and counted visually at ×200 magnification. Data represent mean±SEM of invading cells in 9 random fields of view and are representative of two independent experiments.
Cytokine production. Splenocytes were pooled from at least 3 mice in the various treatment groups and incubated at 1×106 cells/ml with 1×106 cell equivalents/ml of 4T1 tumor cell lysate and 40 U/ml of rhIL-2 for six days. Following restimulation, the splenocytes were collected and plated out at 1×106 cells/ml in triplicate in 24-well tissue culture plates for 48 h. The supernatants were collected and assayed for IFN-γ production by ELISA according to the manufacturer's protocol (eBioscience, San Diego, CA, USA).
Cytotoxicity assay. A standard six-hour 51Cr-release cytotoxicity assay was performed to determine the ability of splenocytes isolated from SM16-treated mice to lyse 4T1 targets (27). 12B1 (H-2d) murine tumor cells were used as irrelevant targets to demonstrate tumor-specific cytotoxicity.

Effect of SM16 treatment on 4T1 primary tumors and metastases. Balb/c mice bearing 9-10 mm2 4T1 tumors were injected i.p. with 40 mg/kg of SM16. SM16 injections were continued daily until day 28. The data represent: A, average tumor size (mm2)±SEM; B, individual tumor sizes (mm2) on day 28; C, number of pulmonary metastases (boxed numbers represent mean±SEM).
Statistical analysis. Statistical significance of differences between data sets was evaluated either by Student's t-test or one-way analysis of variance (ANOVA) including Tukey-Kramer post tests for multiple comparisons using Prism software (GraphPad, San Diego, CA, USA). For all analyses, probability values (P) of <0.05 were considered to indicate significant differences between data sets.
Results
SM16 inhibits TGF-β signaling in 4T1 tumor cells in vitro and 4T1 tumor tissue in vivo. Since the type I TGF-β receptor (ALK5) phosphorylates the transcription factor Smad2 to transduce TGF-β signaling, we measured Smad2 phosphorylation (pSmad2) in 4T1 tumor cells and 4T1 tumors treated with SM16 in order to assess the ability of this drug to inhibit TGF-β signaling. 4T1 tumor cells were treated with different concentrations of SM16 in the presence of exogenous rhTGF-β1 and phosphorylation of Smad2 was analyzed by Western blot. SM16 significantly blocked Smad2 phosphorylation in 4T1 tumor cells in a dose-dependent manner with near complete abrogation of pSmad2 occurring at a concentration of 5 μM (Figure 1A). In addition, 5 μM of SM16 completely inhibited pSmad2 in 4T1 cells in the absence of exogenous TGF-β, indicating that this drug blocks endogenous TGF-β signaling in these cells (Figure 1A).
We also evaluated the ability of SM16 to inhibit TGF-β signaling in established 4T1 tumors. Mice bearing established tumors were injected i.p. with a single dose of either 10 or 40 mg/kg of SM16 and pSmad2 levels were analyzed in primary tumors and lungs bearing metastatic nodules. The data show that a single dose of 10 or 40 mg/kg SM16 profoundly suppressed Smad2 phosphorylation in both primary tumors and lungs bearing a substantial number of metastatic nodules (Figure 1B). This effect was dose dependent as inhibition of pSmad2 in the primary tumor was more effective at a dose of 40 mg/kg (Figure 1B).

Effect of oral SM16 therapy on primary tumors and metastases. Balb/c mice bearing 9-10 mm2 4T1 tumors were transferred to chow formulated with 0.45 g SM16/kg chow. pSmad2 was analyzed in tumors from animals in SM16 diet after 24 h. Serum was collected from animals after 24 h, 36 h, and 28 days on diet and analyzed for SM16 levels. Food and animals were weighed every 4 days and tumor growth was measured 3 times weekly. The data represent: A, pSmad2 levels; B, average weight of mice (g±SEM); C, average SM16 serum levels (μM±SEM); D, average tumor size (mm2±SEM); E, individual tumor sizes on day 28; F, number of pulmonary nodules (boxed numbers represent mean±SEM).
SM16 inhibits TGF-β-induced morphological changes and invasion. Previous work in our laboratory demonstrated that TGF-β plays an important role in migration and invasion of 4T1 tumor cells (16). 4T1 mammary tumor cells normally adhere to glass or plastic surfaces and grow in rounded clumps (Figure 2A). However, when treated with 2 ng/ml rhTGF-β1 for 24 h, the cells assume a spindle cell morphology and demonstrate a dramatic reorganization of the cytoskeleton, characterized by prominent F-actin stress fibers with filopodia and lamellipodia (Figure 2A). This morphological change is completely inhibited by SM16 (Figure 2A). In order to test the ability of SM16 to block TGF-β-mediated invasion, 4T1 cells were treated with the drug and assessed for their ability to invade Matrigel in response to exogenous TGF-β. As shown in Figure 2B, TGF-β stimulated the ability of 4T1 cells to invade Matrigel nearly 3-fold. This effect was completely inhibited by treatment with 5 μM SM16 (Figure 2B).

Effect of SM16 on tumor-specific T-cell immunity. A and B, Spleen cells from SM16-treated animals were restimulated in vitro for 6 days and incubated with 51Cr-labeled 4T1 or 12B1 targets for 6 h. 51Cr release was measured. Data represent mean±SEM of specific lysis; C, Spleen cells from SM16-treated mice were restimulated in vitro for 6 days with 4T1 tumor lysate and IFN-γ production was assayed by ELISA. Data represent mean±SEM of triplicate samples. Data are representative of at least 3 mice/group.
SM16 inhibits primary and metastatic tumor growth. In order to determine the efficacy of SM16 in the treatment of established TGF-β-producing 4T1 tumors, mice bearing 9-10 mm2 established 4T1 tumors were treated with 40 mg/kg of SM16 by daily i.p. injection for 19 days. This dose was chosen based on Smad2 phosphorylation analysis demonstrating complete abrogation of pSmad2 in 4T1 tumor tissue treated with 40 mg/kg of SM16 (Figure 1B). Primary tumors in mice treated with SM16 grew at a significantly slower rate than tumors in mice treated with vehicle alone (p<0.0001) (Figure 3A). At the time of sacrifice on day 28, tumors from SM16-treated mice (average size 88.3±18.7 mm2) were significantly smaller (p<0.01) than those of controls (average size 178.0±13.6 mm2) (Figure 3B). Furthermore, SM16 therapy significantly (p<0.0001) inhibited the formation of pulmonary metastases (Figure 3C). On day 28, mice in the SM16-treated group had an average of 8±3.7 lung nodules compared to 185±34.1 nodules in control mice (Figure 3C). SM16 therapy appeared to be well tolerated and mice displayed no overt signs of drug-related toxicity.
Oral administration of SM16 inhibits primary and metastatic tumor growth. The preceding data demonstrate that SM16 effectively inhibits the growth of primary and metastatic 4T1 mammary tumors when given via daily i.p. injection. In order to make SM16 therapy more clinically desirable, we investigated the efficacy of orally delivered SM16. For this purpose, SM16 was incorporated into standard mouse chow at a dose of 0.45 g SM16/kg chow as described previously (24). In order to determine if SM16 can antagonize TGF-β signaling in tumor tissue when given orally, mice bearing established 4T1 tumors were fed SM16 diet for 24 hours, after which primary tumor tissue was analyzed for pSmad2. Tumors from these animals had substantially reduced levels of phosphorylated Smad2 compared to tumors from animals on the control diet (Figure 4A). To determine the antitumor efficacy of oral SM16, mice bearing established 4T1 tumors (9-10 mm2) were treated with the SM16 diet described above. Each mouse consumed ~3.5 g food per day which is equivalent to a daily SM16 dose of ~1.6 mg/mouse or ~80 mg/kg body weight. Animal weights were comparable in both groups (Figure 4B) and the SM16 diet was well tolerated with no overt signs of drug-related toxicity in the treated animals. Analysis of serum from animals on the SM16 diet revealed detectable levels of SM16 after 24 h on the diet (Figure 4C). Serum SM16 levels increased slightly and reached a peak of ~18.6±1.3 μM after 36 h. This peak level was maintained until the study was terminated on day 28 (Figure 4C). As expected, animals fed normal chow had no detectable SM16 in their serum. Figure 4D shows that SM16 significantly inhibited the growth rate of established 4T1 tumors compared to animals receiving normal chow (p<0.0001) (Figure 4D). At the time of sacrifice on day 28, tumors from the SM16 group had an average tumor size of 58.5±12.9 mm2 versus 139.2±8.7 mm2 for mice on normal chow (Figure 4E). Furthermore, the number of metastatic lung nodules was also significantly reduced in mice receiving the SM16 diet compared to mice receiving normal chow (p<0.0001) (Figure 4F). One out of nine mice fed the SM16 diet experienced complete regression of its primary tumor and was completely tumor free at the end of the study.
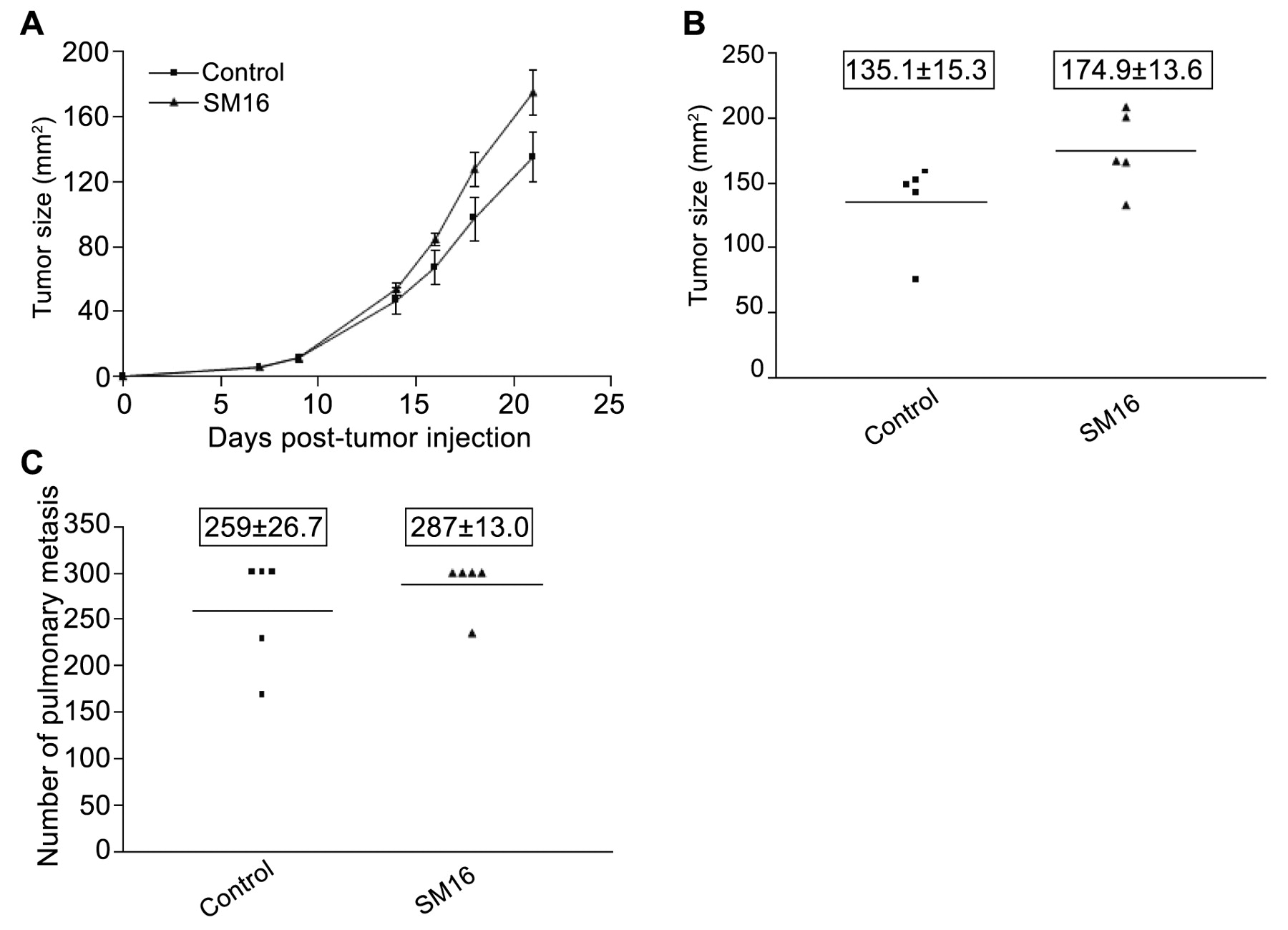
SM16 loses efficacy in immunodeficient mice. C.B17/IcrACCscid mice bearing established 4T1 tumors were fed diet containing 0.45 mg SM16/g chow. Primary tumor sizes were measured and pulmonary metastases were enumerated visually. The data represent: A, B, average tumor size (mm2±SEM); C, number of pulmonary nodules at the time of sacrifice (boxed numbers represent mean±SEM).
The antitumor activity of dietary SM16 has an immune component. To determine if the efficacy of SM16 was related to enhanced antitumor immunity as has previously been reported for other small molecule ALK5 kinase inhibitors (18, 20, 21, 24), splenocytes from tumor-bearing mice on the SM16 diet were restimulated in vitro with 4T1 tumor cell lysate and assessed for cytotoxic activity toward 4T1 targets and IFN-γ production. The data indicate that the cytolytic activity of splenocytes from mice fed SM16 diet was significantly (p<0.001) enhanced compared to splenocytes taken from animals fed the control diet (Figure 5A). This enhanced systemic CTL activity was tumor specific as these effector cells failed to lyse irrelevant 12B1 tumor cells (Figure 5B). As shown in Figure 5C, splenocytes from animals on the SM16 diet produced significantly (p<0.0001) more IFN-γ (122.5±4.8 pg/ml) than their counterparts on the control diet (18.5±0.7 pg/ml).
Since the results described above suggested the involvement of T-cells in the SM16-mediated, antitumor response, we assessed the efficacy of SM16 in tumor-bearing immunodeficient SCID mice. As shown in Figure 6 (A and B), there was no significant difference in primary tumor growth rate or tumor size between SM16-fed and control animals. Interestingly, the tumors grew faster in these animals than in normal immunocompetent BALB/c mice and the study was terminated at day 21 post-tumor injection when animals became moribund. Furthermore, there was no significant difference in the number of pulmonary metastases between animals on the SM16 and control diets (Figure 6C).
Discussion
In this study, we evaluated the effect of the ALK5 kinase inhibitor SM16 on primary tumor growth and metastasis formation in the aggressive 4T1 mammary tumor model that shares similarities with human breast cancer (16, 21, 25). We show that when delivered either via daily i.p. injection or orally through mouse chow, SM16 inhibited the growth of established primary 4T1 tumors and development of pulmonary metastases. This efficacy correlated with reduced Smad2 phosphorylation within primary and metastatic tumor tissue. These findings are consistent with studies in several other tumor models in which ALK5 kinase inhibitors, including SM16 were shown to inhibit primary tumor growth as well as reduce the incidence of tumor metastasis (20-22, 24). In addition, consistent with an earlier report by Suzuki et al. (20), dietary administration of SM16 was safe and resulted in a rapid accumulation of therapeutic levels of SM16 in the serum that were maintained for the duration of the study. The antimetastatic effects of SM16 in our study are particularly striking because even though primary tumors continued to grow, the number of metastatic lung nodules was dramatically reduced in the SM16-treated mice compared to controls. This observation suggests that SM16 therapy may be more effective at treating disseminated disease, where tumor burden is minimal and is in agreement with previous reports by us (16) and others (20-22, 24, 28) that the impact of TGF-β inhibition on metastasis formation is superior to its effect on established primary tumors. While it is clear that SM16 dramatically reduces the incidence of pulmonary metastases in mice bearing established tumors, it is still unclear if the antimetastatic effects of this drug are due to inhibition of the initial seeding of 4T1 cells into the lung, or induction of regression of pre-established metastases. Experiments using SM16 in a residual metastatic disease setting are currently underway to address this question.
Furthermore, the antitumor activity of SM16 was determined to have an immune component. Splenocytes isolated from animals following SM16 treatment demonstrated enhanced IFN-γ production and cytotoxic activity against 4T1 tumor cells compared to splenocytes taken from mice on the control diet. These findings support the hypothesis that tumor-derived TGF-β suppresses antitumor immunity and are consistent with data from Ge et al. (21) showing an enhancement of antitumor immune function following treatment of 4T1 tumors with the ALK5 kinase inhibitor, SD-208. The loss of efficacy of SM16 in immunodeficient SCID mice also suggests the mechanism of action of SM16 requires adaptive immunity. This is consistent with reports by Suzuki et al. (24) and Ge et al. (21) showing that SM16 lost efficacy against the AB12 mesothelioma model in SCID mice and that SD-208 had no effect on the growth of primary and metastatic R3T mammary tumors in athymic nude mice. Taken together, the data indicate that adaptive cellular immunity is critical for the efficacy of SM16 in the 4T1 mammary tumor model.
In addition to augmenting antitumor immunity, TGF-β signaling antagonists have also been shown to control tumor growth through tumor cell autonomous mechanisms such as blocking the EMT and invasion. Although we demonstrate that SM16 blocks TGF-β-induced 4T1 cell invasion in vitro, this mechanism does not appear to play a major role in the antimetastatic effects of this drug in vivo, as SM16 had no effect on the formation of lung metastases in immunodeficient SCID mice. This finding is consistent with a report by Ge et al. (21) who showed that SD-208 had no effect on R3T mammary tumor growth and metastasis formation in athymic nude mice despite its ability to inhibit R3T EMT and invasion in vitro. However, our data conflict with a report by Bandyopadhyay et al. (29) showing that a small molecule ALK5 kinase inhibitor reduced the ability of human MDA-MB-435 xenografts to form spontaneous and experimental lung metastases. The disparity in these findings may be attributed to the different tumor models (mouse versus human) used in both studies.
Data from studies in several different human tumor xenograft models have shown that the efficacy of a number of TGF-β inhibitors, including the pan-TGF-β neutralizing antibody 1D11, a soluble TGF-β receptor type III, and a dominant negative TGF-β receptor type II, also involve an inhibition of angiogenesis (30-33). However, the failure of SM16 to inhibit 4T1 tumor growth in immunodeficient mice in our studies suggests that suppression of angiogenesis may not play a major role in the efficacy of SM16 in the 4T1 tumor model. Similarly, Uhl et al. (20) were unable to demonstrate a significant difference in microvessel density in SMA-560 gliomas treated with SD-208. Interestingly, Ge et al. (21) have shown that SD-208 treatment does cause a reduction in microvessel density in R3T mammary tumors, however, the importance of this mechanism in the efficacy of this inhibitor is controversial as SD-208 had no effect on the growth of R3T tumors in the absence of functional T-cell immunity in athymic nude mice. Ultimately, these discrepancies may be due to differences in the tumor models and TGF-β inhibition strategies employed in each of these studies.
Conclusion
To the best of our knowledge, this is the first published report demonstrating the therapeutic efficacy of SM16 on established primary and metastatic mammary tumors. Unlike the study of Ge et al. (21) using another ALK5 kinase inhibitor in which treatment was initiated in a prevention setting (1 day post-tumor injection), our study focused on the treatment of established primary disease as well as spontaneous metastases. We show that SM16 is effective in limiting primary tumor growth and abrogating spontaneous lung metastases in the more difficult to treat established disease setting. Similar to what has been reported by others with related ALK5 inhibitors in different tumor models (20, 21, 24), the efficacy of SM16 against 4T1 tumors is dependent on an immune-mediated mechanism. Furthermore, SM16 whether delivered via daily i.p. injection, or orally in feed is safe and does not result in any of the adverse side-effects seen with genetic methods utilized to inhibit TGF-β signal transduction (28, 34, 35). More importantly, the ability of SM16 to modulate endogenous antitumor immunity provides a strong rationale for using it in combination with immunotherapy in the treatment of metastatic breast cancer. Experiments are currently underway to investigate this approach.
Acknowledgements and Disclosures
Grant Support: 1 RO1 CA94111-02 and P30-CA23074 from the NIH and W81XWH-06-1-0438 from the DOD. We thank Ellen Rohde and Cheryl Black for excellent bioanalytical mass spectrometry analysis for these studies. We would also like to thank the Tissue Acquisition and Cellular/Molecular Analysis Shared Service Core (TACMASS) at the Arizona Cancer Center, supported by NIH grant CA23074 for the generation of Immunohistochemical data. LEL, H-KC, W-CL, and XZ are Biogen Idec employees and shareholders. Biogen Idec is not developing SM16 for clinical use.
- Received September 19, 2008.
- Revision received December 17, 2008.
- Accepted March 11, 2009.
- Copyright© 2009 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- In silico trials predict that combination strategies for enhancing vesicular stomatitis oncolytic virus are determined by tumor aggressivity
- Urethane-induced Mammary Carcinogenesis Susceptibility in Transgenic Mice Expressing a Dominant-negative TGF-{beta} Type II Receptor
- The Marine-Derived Oligosaccharide Sulfate MS80, a Novel Transforming Growth Factor {beta}1 Inhibitor, Reverses Epithelial Mesenchymal Transition Induced by Transforming Growth Factor-{beta}1 and Suppresses Tumor Metastasis
- STAT3 Signaling Is Required for Optimal Regression of Large Established Tumors in Mice Treated with Anti-OX40 and TGF{beta} Receptor Blockade
- TGF{beta} Inhibition Prior to Hypofractionated Radiation Enhances Efficacy in Preclinical Models
- TGF-{beta}/SMAD/GLI2 Signaling Axis in Cancer Progression and Metastasis