


Abstract
Etoposide [4′-demethylepipodophyllotoxin-9-(4,6-O-ethylidene)-β-D-glucopyranoside] is a substrate for P-glycoprotein (P-gp) and cytochrome P450 (CYP) 3A. This study was designed to investigate the effects of quercetin (3,5,7,3′,4′-pentahydroxyflavanone), a P-gp and CYP3A inhibitor, on the pharmacokinetics of etoposide in rats. Etoposide was administered to rats orally (9 mg/kg) or i.v. (3 mg/kg) without or with quercetin (1, 5 or 15 mg/kg). The plasma concentration of etoposide was determined by high performance liquid chromatography (HPLC) equipped with a fluorescence detector. In the presence of quercetin, the pharmacokinetic parameters of etoposide were significantly altered in the oral group, but not in the i.v. group. The presence of quercetin significantly (5 mg/kg, p<0.05; 15 mg/kg, p<0.01) increased the area under the plasma concentration-time curve (AUC) of orally administered etoposide from 43.0 or 53.2% . The presence of 5 or 15 mg/kg of quercetin significantly (p<0.05) decreased the total body clearance (CL/F) of oral etoposide. Consequently, compared to the control group (8.87%), the presence of quercetin significantly (5 mg/kg, p<0.05; 15 mg/kg, p<0.01) increased the absolute bioavailability (AB) of etoposide to 12.7 or 13.6% . The enhanced oral bioavailability of etoposide by quercetin could mainly be due to inhibition of P-gp-mediated efflux and CYP3A-catalyzed metabolism in the intestine by quercetin. The dosage regimen of etoposide in cancer therapy should take drug interaction into consideration when etoposide is administered with quercetin or dietary supplements containing quercetin.
Etoposide [4′-demethylepipodophyllotoxin-9-(4,6-O-ethylidene)-β-D-glucopyranoside] is used in the treatment of a wide range of malignancies such as lung cancer, acute leukemia and lymphoma (1). Etoposide is converted to an O-demethylated metabolite by the hepatic microsomal cytochrome P450 (CYP) in rat (2) and human (3) liver microsomes. Demethylation of etoposide is mediated mainly by CYP3A4 and to a minor extent by CYP1A2 and 2E1 (4). Approximately 20-35% of the etoposide dose is excreted unchanged in the urine and ~2% in the bile (3). P-glycoprotein (P-gp) has been shown to affect the absorption of etoposide in the intestine (5, 6). Orally administered etoposide exhibits variable bioavailability with a range of 25-75% (1). CYP3A4 and P-gp in the intestine could be among the main contributors to the erratic bioavailability. Thus, the concomitant administration of CYP3A4 and P-gp inhibitors might improve the oral bioavailability of etoposide.
The flavonol quercetin (3,5,7,3′,4′-pentahydroxyflavanone) is one of the most prevalent flavonoids in the diet. It is present in fruits, vegetables and beverages mainly as glucosides, and the highest content is found in onions, apples and red wine (7, 8). Quercetin is nontoxic and has a variety of biological effects such as anticancer (9), antioxidant (10), antiviral (11), antiulcer (12) and antiallergic (13) activities. Daily dietary intake of quercetin ranges from 4 to 68 mg based on epidemiological studies in the U.S.A., Europe and Asia (8, 14-16). It has been reported that quercetin competitively inhibited the metabolizing enzyme, CYP3A4 (17-20) and the members of the multidrug resistance (MDR) family, P-gp, multidrug resistance-associated proteins 1 (MRP1) and the breast cancer resistance protein (BCRP) (21). Quercetin and etoposide could be prescribed for the treatment or prevention of cancer as a combination therapy. Therefore, this study investigated the effects of quercetin on the bioavailability and pharmacokinetics of etoposide after oral or i.v. administration etoposide in rats.
Materials and Methods
Materials. Etoposide, quercetin and podophyllotoxin (an internal standard for high performance liquid chromatography, HPLC analysis of etoposide) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Injectable etoposide (20 mg/ml) was purchased from Boryung Chemical Co. (Seoul, Republic of Korea). Methanol and tert-butylmethylether were acquired from the Merck Co. (Darmstadt, Germany). The other chemicals were of reagent grade or HPLC grade.

Chromatogram of blank rat plasma (A) and plasma spiked with etoposide and the internal standard, podophyllotoxin (B).
Animal experiments. Seven- to 8-week-old male Sprague-Dawley rats weighing 270-300 g were purchased from Dae Han Laboratory Animal Research Co. (Choongbuk, Republic of Korea) and given free access to commercial rat chow (No. 322-7-1, Superfeed Co., Gangwon, Republic of Korea) and tap water ad libitum. The animals were housed, four or five per cage, in laminar flow cages maintained at 22±2°C and 50-60% relative humidity, under a 12:12 h light-dark cycle. The experiments began after acclimation to these conditions for at least one week. The protocols of the animal studies were approved by Animal Care Committee of Chosun University (Gwangju, Republic of Korea).
Drug administration. The rats were divided into four groups (n=6) for each of the oral and i.v. etoposide administration routes (9 mg/kg for oral; 3 mg/kg for intravenous) with 0 (control), 1, 5 or 15 mg/kg of orally administered quercetin. The rats were fasted for at least 24 h prior to beginning the experiments and had free access to tap water. Each animal was lightly anaesthetized with ether, and the femoral artery and vein were cannulated using polyethylene tubing (SP45; i.d., 0.58 mm, o.d., 0.96 mm; Natsume Seisakusho Co. Ltd., Tokyo, Japan) for blood sampling and i.v. administration. Etoposide (20 mg/ml injectable solution diluted in 0.9% NaCl-injectable solution) at a dose of 3 mg/kg was injected (total injection volume, 1.5 ml/kg) over 1 min via the femoral vein 30 min after the administration of 0 (control), 1, 5 or 15 mg/kg of quercetin (dissolved in distilled water, homogenized at 36°C for 30 min; total oral volume, 3.0 ml/kg) through a feeding tube. Blood samples (0.2 ml) were collected into heparinized tubes via the femoral artery at 0 (as a control), 0.017 (at the end of injection), 0.1, 0.25, 0.5, 0.75, 1, 2, 4, 6 and 8 h the after the intravenous injection of etoposide. The blood samples were immediately centrifuged for 5 min at 13,000 rpm and a 0.1 ml aliquot of plasma was stored in a freezer at -40°C until HPLC analysis. Etoposide (the same solution as used for i.v. administration) was administered orally at a dose of 9 mg/kg (total oral volume, 3.0 ml/kg) without or with 1, 5 or 15 mg/kg of orally administered quercetin. Blood samples (0.2 ml) were collected into heparinized tubes via the femoral artery at 0 (as a control), 0.1, 0.25, 0.5, 0.75, 1, 2, 4, 6, 8 and 10 h after the oral administration of etoposide. The other procedures were similar to those in the i.v. studies.
HPLC analysis. The plasma concentrations of etoposide were determined by the HPLC assay reported by Liliemark et al. (22) and Manouilov et al. (23) with slight modification. Briefly, 50 μl of podophyllotoxin (50 ng/ml dissolved in methanol) and 1.2 ml of tert-butylmethylether were mixed with a 0.1 ml aliquot of the plasma sample in a 2.0 ml polypropylene microtube (Axygen Scientific Co., Calif., USA). The resulting mixture was mixed vigorously with a vortex-mixer (Scientific Industries Co., NY, USA) for 1 min and centrifuged at 13,000 rpm for 10 min with a high-speed micro centrifuge (Hitachi Co., Tokyo, Japan). A 1.1 ml aliquot of the upper layer was transferred to another clean microtube and evaporated under nitrogen gas at 38°C in an MG 2100 Eyela dry thermo bath (Rikakikai Co., Tokyo, Japan). The residue was dissolved in 0.2 ml of 50% methanol in deionized water, and a 50 μl aliquot of the solution was injected into the HPLC system.
The HPLC was equipped with a Waters 1515 isocratic HPLC Pump, a Waters 717 plus autosampler and a WatersTM 474 scanning fluorescence detector (Waters Co., Milford, MA, USA). Data were acquired and processed with breeze™ Software (Version 3.2) (Waters Co.). Chromatographic separations were achieved using a Symmetry® C18 column (4.6×150 mm, 5 μm, Waters Co.) and a μBondapak™ C18 HPLC Precolumn (10 μm, Waters Co.). The mobile phase consisted of methanol-deionized water-acetic acid (50:50:0.5, v/v/v) and run at a flow-rate of 1.0 ml/min. Chromatography was performed at 30°C, which was set by a HPLC column temperature controller (Phenomenex Inc., CA, USA). The fluorescence detector was operated at an excitation wavelength of 230 nm with an emission wavelength of 330 nm. Podophyllotoxin and etoposide eluted with retention times of 5.4 and 11.1 min (Figure 1), respectively. The presence of quercetin did not interfere with the detection of etoposide and podophyllotoxin. The detection limit of etoposide in plasma was 10 ng/ml. The intra- and inter-day variation coefficients of etoposide in the plasma samples were below 13.6% .
Pharmacokinetic analysis. The plasma concentration data were analyzed by noncompartmental methods using WinNonlin software version 4.1 (Pharsight Corporation, Mountain View, CA, USA). The elimination rate constant (Kel) was calculated by log-linear regression of etoposide concentration data during the elimination phase and the terminal half-life (t1/2) was calculated as 0.693/Kel. The peak concentration (Cmax) of etoposide in the plasma and the time to reach Cmax (tmax) were obtained by visual inspection of the data from the concentration-time curve. The area under the plasma concentration-time curve (AUC) from time zero to the time of last measured concentration (Clast) was calculated by the linear trapezoidal rule. The AUC zero to infinite (AUC0-∞) was obtained by adding AUC0-t and the extrapolated area determined by Clast/Kel. The total body clearances of i.v. (CLt) and oral (CL/F) etoposide were calculated from the quotient of the dose (D) and the AUC0-∞ of the i.v. and oral routes. The absolute bioavailability (AB) was calculated by AUCoral/AUCi.v. × Dosei.v./Doseoral × 100. The relative bioavailability (RB) was calculated by AUCcoadmin/AUCcontrol × 100.

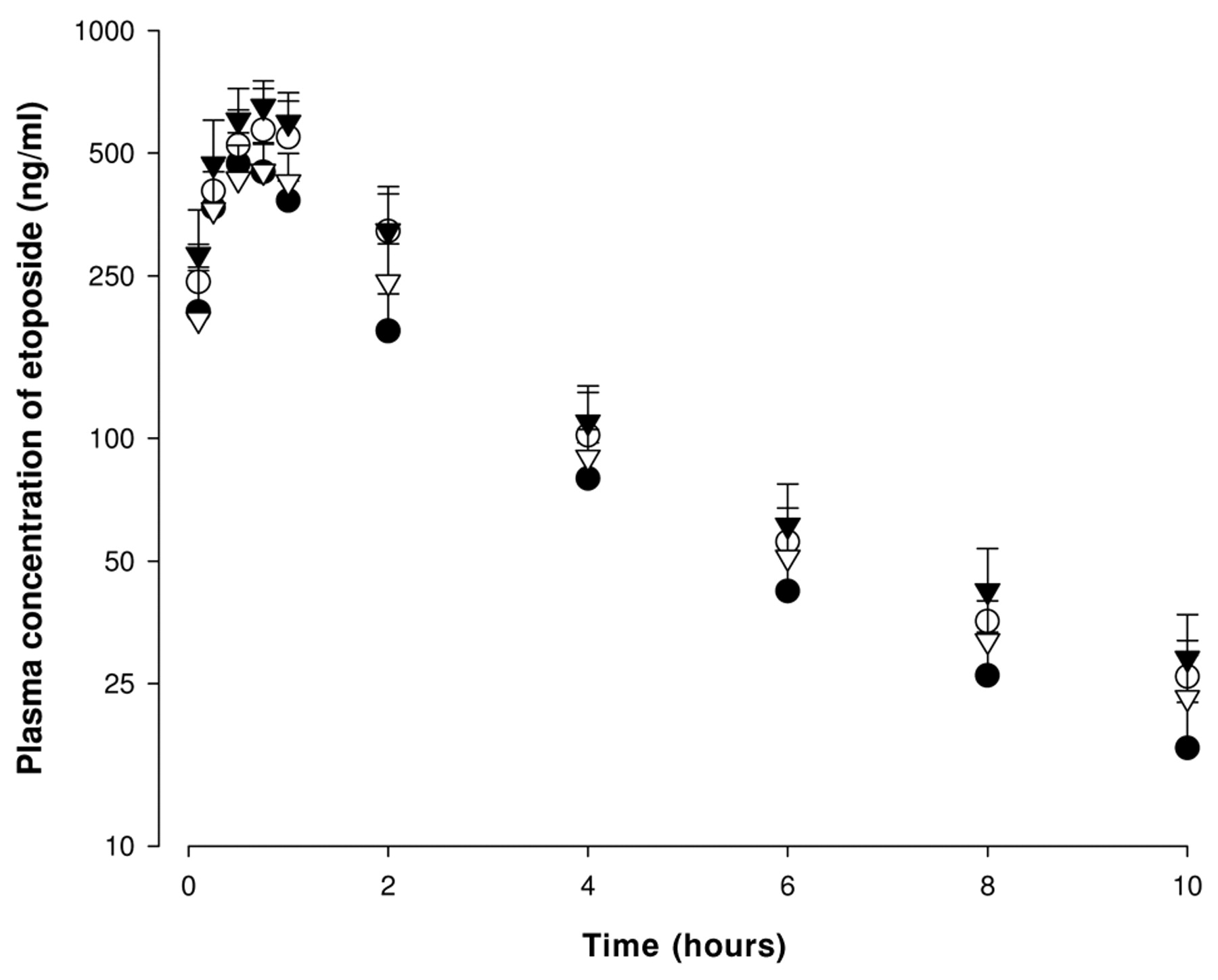
Mean plasma concentration-time profiles of etoposide after oral administration of etoposide at a dose of 9 mg/kg to rats in the absence or presence of quercetin at doses of 1, 5 or 15 mg/kg (Mean±S.D., n=6). Bars represent the standard deviation. (●) without quercetin or with (▿) 1 mg/kg, (○) 5 mg/kg and (▾) 15 mg/kg quercetin.
Statistical analysis. The pharmacokinetic parameters were compared using a one-way ANOVA, followed by a posteriori testing with the Dunnett correction. Differences were considered significant at a level of p<0.05. All the data are expressed in terms of mean±S.D.
Results
The mean arterial plasma concentration-time profiles of etoposide following oral administration in the presence or absence of quercetin are illustrated in Figure 2, and the pharmacokinetic parameters of etoposide are summarized in Table I. Compared with the control group (etoposide alone), the presence of quercetin significantly (5 mg/kg, p<0.05; 15 mg/kg, p<0.01) increased the area under the plasma concentration-time curve (AUC) of etoposide by 43.0 or 53.2%, and the presence of quercetin (5 or 15 mg/kg) significantly (p<0.05) increased the peak plasma concentration (Cmax) of etoposide by 21.1 or 39.0%, repsectively. The presence of quercetin (5 or 15 mg/kg) significantly (p<0.05) decreased the total body clearance (CL/F) of etoposide by 27.6 or 33.7%, respectively. Consequently, compared to the control group (8.87%), the presence of quercetin significantly (5 mg/kg, p<0.05; 15 mg/kg, p<0.01) increased the AB of etoposide to 12.7 or 13.6%, and RB of etoposide was increased by 1.43- or 1.53-fold above that of the control group, respectively. There were no significant changes in the time to reach the maximum plasma concentration (Tmax), the elimination constant (Kel) and the terminal half-life (t1/2) of etoposide. Although 1 mg/kg of quercetin decreased the CL/F and increased the AUC of etoposide, these changes were not statistically significant.

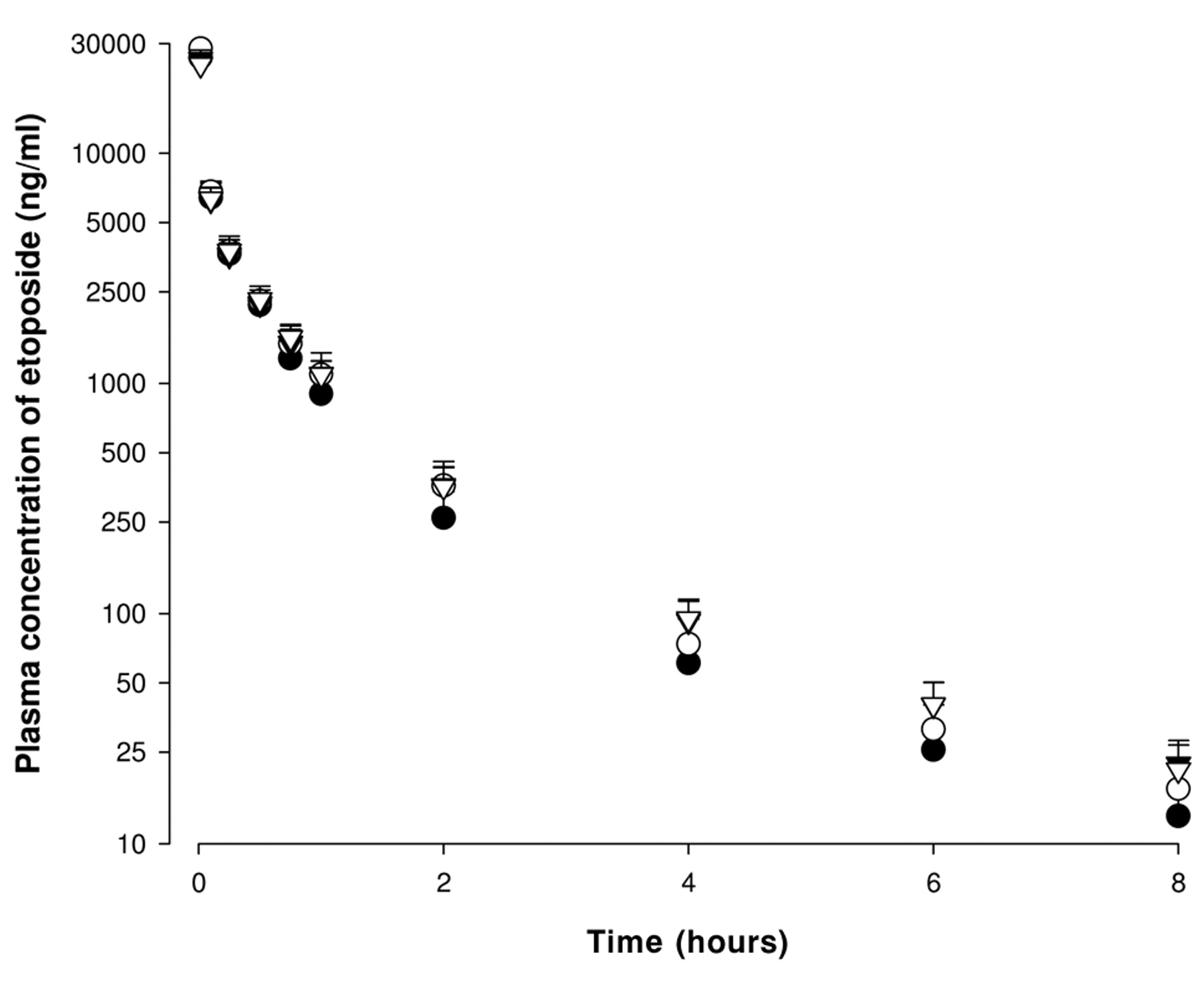
Mean plasma concentration-time profiles of etoposide after intravenous administration of etoposide at a dose of 3 mg/kg to rats in the absence or presence of quercetin at doses of 1, 5 or 15 mg/kg (Mean±S.D., n=6). Bars represent the standard deviation. (●) without quercetin or with (○) 1 mg/kg, (▿) 5 mg/kg and (▾) 15 mg/kg quercetin.
The mean arterial plasma concentration-time profiles of etoposide following i.v. administration of 3 mg/kg etoposide in the absence or presence of 1, 5 or 15 mg/kg of quercetin are shown in Figure 3, and the pharmacokinetic parameters of the i.v. etoposide are listed in Table II. Although quercetin increased the AUC and decreased the CLt of etoposide, these changes were not statistically significant. The other parameters such as Kel and t1/2 were not altered significantly.
Discussion
Following the oral administration of etoposide, quercetin (5 or 15 mg/kg) significantly increased the AUC of etoposide and significantly reduced the CL/F of etoposide (Table I), but quercetin did not significantly alter the pharmacokinetics of i.v. administered etoposide. These results were similar to our earlier work where morin (15 mg/kg) increased the AUC and Cmax of orally administered etoposide compared with the control, mainly due to inhibition of CYP enzymes and P-gp in the intestine (24). In the previous study, infusion of cyclosporine A (1 mg/h) in rats treated with an i.v. bolus of etoposide decreased the plasma clearance (5.4±2.1 vs. 9.3±2.4 ml/min), and increased the plasma and tissue concentrations of etoposide, although not significantly (25). Lo and Huang (26) also reported that quercetin increased etoposide absorption in everted sacs of the jejunum or ileum. Therefore, the increased oral bioavailability of etoposide (at 5 or 15 mg/kg of quercetin) might have been caused by competitive inhibition of P-gp and CYP3A mainly in the intestine by quercetin. Interestingly, these results were coincident with another study in our laboratory (27) in which coadministration of tamoxifen with 2.5 and 7.5 mg/kg of quercetin in rats significantly increased AUC and Cmax of orally administered tamoxifen compared to the control group (tamoxifen alone). This result was similar to a previous study in which a low dose (2 mg/kg) of quercetin did not significantly change the bioavailability of paclitaxel prodrug after oral administration in rats (28). CYP3A and P-gp in the liver and kidney were not markedly inhibited by the quercetin in the i.v. administration group, therefore, quercetin did not significantly increase the bioavailability of etoposide. Although further characterization is needed, quercetin may modulate the intestinal extraction of P-gp and/or CYP substrates in a dose-dependent manner.
Pharmacokinetic parameters of etoposide after oral administration at a dose of 9 mg/kg to rats in the absence (control) or presence of quercetin at doses of 1, 5 or 15 mg/kg (Mean±S.D., n=6).
Pharmacokinetic parameters of etoposide after intravenous administration at a dose of 3 mg/kg to rats in the absence (control) or presence of quercetin at doses of 1, 5 or 15 mg/kg (Mean±S.D., n=6).
Collectively, the coadministration of quercetin might be an approach to improve the erratic bioavailability of etoposide. Further studies in humans are needed to elucidate the clinical implication of these findings in rats.
Conclusion
The enhanced oral bioavailability of etoposide might be attributed to inhibition of P-gp and CYP3A by quercetin in the intestine of rats. The dosage regimen of etoposide in cancer therapy should take drug interaction into consideration when etoposide is administered with quercetin or dietary supplements containing quercetin.
- Received May 5, 2008.
- Revision received January 14, 2009.
- Accepted February 12, 2009.
- Copyright© 2009 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}