


Abstract
The objective of this study was to find whether the peptide LRMK linked to the N-terminus of HER-2, 774-788 and shorter peptides create a T helper antigen, which can replace all functional HER-2 peptides in a cancer vaccine. Of the 6 LRMK-HER-2 peptides tested in the presence of IL-12, AE-37, AE-38 and AE-39 induced higher IFN-γ in PBMCs from 4 healthy donors than the other peptides. AE-37 and AE-39 contained the immunogenic HER-2 peptide, p776 (774-788), while AE-38 contained the truncated HER-2 peptide G89 (777-788). The free, unprotected HER-2 peptide G89 (777-789) activated IFN-γ production in PBMCs from another BRC patient. Responses to free p776 and F7 could not be tested. Three out of 11 patients responded strongly only to AE-37, while 3 of 10 patients responded strongly only to G89. One responded only to AE-39. All 3 patients diagnosed with DICS of mixed HERhi, ER+ PR+ type responded to AE-37. Three out of 4 patients diagnosed with luminal cancer and one HERhi, ER- PR- BRC patient responded only to G89. AE-37, at low concentration, helped to expand more E75-TCRHi+Med cells than the negative control AE-47, for IFN-γ induction AE-47, but was a weaker helper than AE-47 at higher concentrations, and eliminated E75-TCRHi cells. The strongest and most consistent effect of AE-37 was the elimination of CD4 Hi CD25 Hi cells. When LRMK is linked to AE-37, the side-chains of L P-10 and R P-9 are positioned away from MHC; LRMK forms a bi-strophynx (rotating-double-hook-like) structure when attached to AE-37 and the minimum HER-2 peptide in the peptide-binding groove (PBG). K P-8 anchors the bi-strophynx to HLA-DR outside the PBG in a novel site DRα, 49-52. The HER-2 amino acids, Y P-1 and R P3 point towards TCR; R P-9 and M P-8 can contact the TCRVα1. The positions of K P-8 in AE-37 and Ava (εV) P-8 in AE-39 modulated immunogenicity of p776. It is unknown whether LRMK adds TCR contact points to the minimal HLA-DR-bound HER-2 peptide (780-788) or activates T-cells of other specificities to produce cytokines and die. Preferential activation of Th1 cells in distinct individuals by AE-37, G89 and AE-39 indicates that cancer vaccines will benefit from correct individual and disease-associated help. Emergence of distinct daughters of luminal and myoepithelial BRC stem cells during metastasis through “de-differentiation” and “reverse differentiation” of drug-resistant cancer requires personalized vaccines, which use an optimal-helper antigen.
Abbreviations: P, position of the amino acid in the peptide; P1, position of the first amino acid anchored in MHC-II-peptide binding groove; P-1, P-2, etc, position of amino acids outside the peptide binding groove; PBG, peptide-binding groove; εV, Ava, ε-aminovaleric acid; BRC, breast cancer; G89, HER-2 (777-789); AE37, LRMK-HER-2(774-788); AE39, LRMK-Ava-HER-2(774-788); AE47, LRMK-Ava-HER-2(776-788); TCR, T-cell receptor; SAG, superantigen; Å, angstrom.
Sequences of HER-2 peptides aligned at P1, their main HLA-DR-binding anchor.
This paper reports our findings regarding the activation of peripheral blood mononuclear cells (PBMCs) from the same breast cancer (BRC) patient by free G89 and ‘protected’-Ii-key (LRMK)-linked p776 and F7. p776 induced Th1 responses in p776/HER-2 vaccinated BRC patients (1-3). F7 activated PBMC from ovarian cancer patients in vitro and BRC-patients (4, 5). In vitro, G89 activated CD4+ cells from lymph nodes and PBMCs of BRC patients and produced more interferon (IFN)-γ than interleukin (IL)-4 and IL-10. G89 is considered as Th0/Th1 tumor antigen (4, 5). p776 is the peptide identified by Dr. Disis and Cheever's laboratories, HER-2(774-788, or 776-790) (6, 7). The two peptides identified by Dr. Ioannides laboratory are HER-2(776-788) [F7] and HER-2(777-789) [G89] (4, 5). F7 and p776 have the same C-terminus, while G89 has an additional amino acid (threonine) at the C-terminus. F7 is shorter than p776 with the amino acids (Gly-Val).
The study was requested to: (i) address the hypothesis that the N-terminus of peptides bound to MHC-II is significant for recognition and activation of T-cells, and (ii) identify the optimal position of extension of HER-2 peptide chain with LRMK alone or LRMK-εV to create an immunogenic compound. The N-terminus extended with LRMK is the terminus of the flanking/outside chain of the HLA-DR-bound HER-2 peptide and not the N-terminus of the HLA-DR-PBG 9 amino acids' long HER-2 peptide. The predicted HLA-DR-PBG peptide was the same in all compounds.
We corrected the sequence of G89. G89 was listed in reports on AE peptides as extended or truncated with two amino acids added or deleted. Sequences are presented with numbers above each amino acid. They start from P1 = the first HLA-DR anchor of the minimum HER-2-peptide bound to the HLA-DR-PBG (Tables I and II).
Materials and Methods
Patients. PBMCs were isolated from 26 BRC patients and 9 healthy donors. All patients and healthy volunteers signed informed consent to participate in the study, under research protocols approved by MDACC.
Peptides and reagents. Ii-Key-peptides were reported elsewhere (8-12). LRMK-Ava was found by Dr. Ioannides to be the less toxic and the more activating from a group of 4-5 LRMK-containing peptides synthesized as one peptide with G89 (13). Peptides had N- and C-termini “protected”. Mass spectrometry sequencing, curiously, shows only one peak instead of the expected 3 to 5. The content of Ava was not determined. We are not aware of the composition of AE-peptides. Free p776, F7 and LRMK-Ava-G89 were not provided. G89 and E75 (369-377) were synthesized as free peptides and purified by the Core facility of MDACC (5, 14). All reagents were from BD PharMingen (San Diego, CA, USA).
Immunodominance. Prior to surgery, or at least 3 weeks after chemotherapy, 40 ml (5 green-cap tubes or less) of blood were drawn. The blood was delivered immediately to the laboratory. PBMCs were separated, stained for HLA-A2 expression and cultured immediately by Drs. Kawano and Efferson to minimize losses from freezing. All peptides (Table II) were added at the same time to PBMCs at 25 μg/ml following Dr. Ioannides' procedures to increase sensitivity of detection (15-21). ELISPOT was performed according to Cellular Technology Limited (CTL, Cleveland, OH, USA) for 6-7 patients. Spots were counted by CTL. Quantification of E75-TCR+ CD8+ cells followed our methods as reported elsewhere (5, 20, 21). Analysis of quantity, quality, help and kinetics of T-cell responses to AE peptides determined by us is reported elsewhere with the names of peptides changed to A, B, C, D, E and F (Murray et al. submitted, 2008). G89 and AE-37 were designated immunodominant when they induced 2 times more IFN-γ than the other peptides; they were designated as co-dominant when the amount of IFN-γ induced in all peptides was within a 20% range. Immunodominance was defined from ELISPOT only when ELISA results were not available.
Optimal binding peptide (blue) to each HLA-DR molecule from HER-2, 769-788.
Results
Structural analysis and functional implications of the HLA-DR: G89, F7, p776 and LRMK-p776 complexes. It was proposed that addition of the 4 amino acids of human Clip (77-80, LRMK; Table I), to the N-terminus of an immunogenic HER-2 peptide would enhance binding of the longer “fusion peptide” to MHC-II as it does for Clip (8-12). Stronger MHC-II-bound peptides activate more cells than natural peptides. Our findings of the enhancement of activation by G89 by the shortest Clip (77-80)-linked to εV were interpreted to support the increase in G89 immunogenicity by an increase in affinity for HLA-DR of the linked {Clip (77-80)-HER-2 (777-789)} peptide.
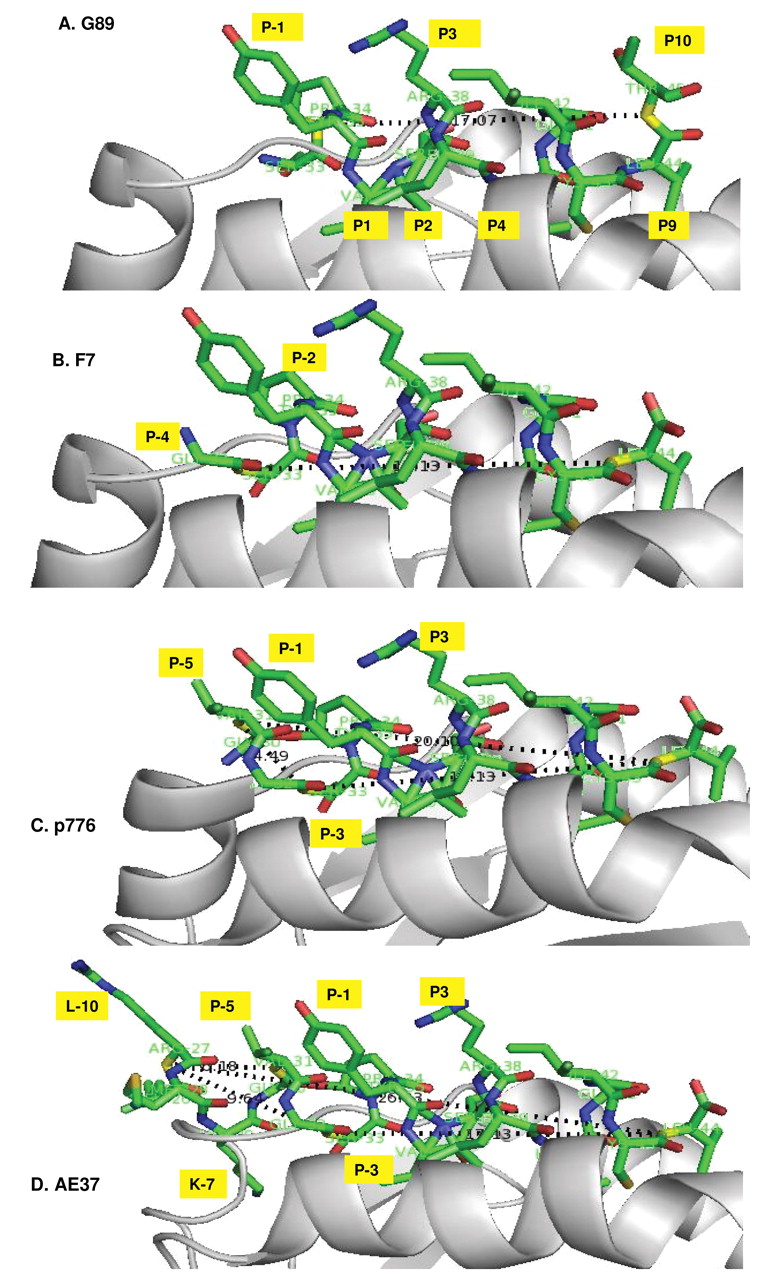
Models of HER-2 peptide bound to HLA-DR1. (G89, F7, p776 and AE-37) show the binding site of LRMK of AE-37 to HLA-DR. Technical details in references 43 and 44. Positions of the amino acids in the peptides are as listed in Table I. The lysine of LRMK binds the strand connecting DR chain with β-sheet plane underneath the peptide-binding groove and destabilize the N-terminal helices of DR-chain. Model constructed with the PDB files 1SJE and 1SJH, for HLA-DR1-Gag complex from reference 43. The TCR is specific for Gag-peptide. The distance between the first and last peptide bonds is around 20Å in G89 and F7 and 40Å in AE-37.
The concept predicts Clip (77-80) located outside the peptide-binding groove (PBG) enhances binding of the fused peptide to an allosteric site. Clip (91-99, MRMATPLLM) binds the PBG of HLA-DR (8-12, 22, 23). Clip (81-90) is N-terminal to Clip (91-99), as is Clip (77-80) to Clip (81-90). This means that the allosteric site is 10 to 14 amino acids away from the P1 anchor amino acid (Met) of the DR-PBG. CLIP (91-99) binds with high affinity to DR3, and more weakly to DR5, DR4w4, DR4w14c and DR52a (22, 23). We could not find reports of allosteric sites on MHC α- and β-chains located 10 to 14 amino acids away from the PBG.
The three amino acids proximal to Clip (91-99) = {Clip [(88-90), VSK]} are solvent-exposed and lift from the α-peptide plane. The rest of Clip (81-87, LPKPPKP) is proline rich. Its position has not been determined but authors have proposed that it is disorganized (22). It was proposed that CLIP (77-90) bends and binds to an allosteric site on HLA-DR, through Clip ((77-80) (LRMK)). In AE peptides, LRMK, together with flanking HER-2 peptide, are shorter than the 10-14 amino acids required for Ii-key to reach its natural allosteric site (Table I). Therefore, we searched for an alternative HLA-DR-binding site for LRMK.
The minimum peptide bound in the PBG of HLA-DR is 9 amino acids long (24, 25; Table I). The minimum predicted HLA-DR bound HER-2 peptide is 780-788: VSRLLGICL (26-27). F7 (HER-2, 776-788, GSPYVSRLLGICL), is the natural processed peptide. F7 contains the flanking/outside peptide, GSPY (28). All the “helper” HER-2 peptides discovered and evaluated independently by Drs. Ioannides, Cheever and Disis, contain the minimum HER-2 peptide (780-788). We asked whether the side chains of the LRMK “outside” amino acids of the AE peptides are positioned towards HLA-DR-α/β-chains - an indication that LRMK bind HLA-DR or they are positioned away from MHC - an indication that LRMK bind TCR.
Drs. Ioannides and Li built molecular models to identify peptide-induced changes in energy of binding in DR (29). The flanking/outside-chain is short in G89 (=SPY) longer in F7 (=GSPY) and longest in p776 (=GVGSPY) (Figure 1). When LRMK is added, the length of the entire outside-chain increases to 10 amino acids, LRMKGVGSPY in AE-37, and 11 amino acids, LRMK-εV-GVGSPY, in AE-39. The HER-2 flanking/outside-chain but not LRMK increased the affinity of AE peptides for HLA-DR (30).
AE-47 (F7) and AE-39 (p776) are extended with εV positioned before LRMK. εV creates an N-δ to C-α peptide bond with Lys (K) in LRMK. The N-δ to C-α -peptide bond should twist the LRMK or allow it more flexibility. εV moves LRMK away from the PBG in AE-39. The shorter HER-2 flanking/outside-chain of AE-47 brings LRMK closer to PBG. εV-Containing AE-38 and AE-48 move LRMK closer, or farther away from the PBG. The distance of LRMK from the HLA-DR-PBG increase in this order: AE-38 (closest)->AE-47->AE-48 = AE-37->AE-39 (Figure 1).
We found a novel HLA-DR-anchored structure of AE-37. The Leu P-10 side chain and the positively charged side chain of R P-9 in AE-37 are oriented away from MHC. The side chain (SCH3) of M P-8 has no specific orientation [Figures 1 (AE-37) and 2]. The K P-7 side chain is deep in HLA-DR and disorganize the two first helices of HLA-DR. This effect is unique compared with AE-47 (29). Therefore, in AE-37, LRMK looks like a two-hook-stirrer. We named this structure bi-strophynx (strophynx, στροφυνξ = Greek word for rotator), and Sanchaji = Chinese for a three-spike-fork ancient Chinese weapon.

Bi-strophynx orientation of LRMK. A, Orientation of HER-2 amino acids outside the HLA-DR1 peptide-binding groove extended with LRMK in AE-47. B, The electrostatic map of the LRMK (structure made with the Swiss-protein SPDBV program) indicate that this peptide can contact both HLA-DR and TCR. The “Sanchaji” -Bi-strophynx model of LRMK made from Figure 1. See also Figure 7.
R P3, in the minimum HER-2-PBG of G89, F7 and p776 points towards TCR. Y P-3, in the flanking/outside chain, can form a secondary contact with TCR. T P-10 in G89 creates a third TCR contact by forming hydrogen bonds with TCR. T P-10 may enhance immunogenicity.
Val P-5 can provide additional TCR contact in AE-37 and AE-39 but not in AE-47 and G89. R P-9 from Clip forms the strophynx, which points to both TCR and MHC (Figure 2). The strophynx is absent from p776. In all other AE-peptides, εV addition, and amino acid deletions, in HER-2 flanking/outside chain from the PBG changed position of the strophynx and of V P-5.
To summarize, the R P3 in minimum HER-2 peptide and the flanking/outside Y P-1 points toward TCR. R P-3 and Y P-1 in AE-37 can engage the same TCR as AE-39, AE-47 and G89. R P-9 and V P-5, in AE-37, will contact the CDRα1 domain of the HER-2 specific TCR, or the CDR3-α domain of a different TCR. When AE-37 binds its HER-2 peptide to HLA-DR-PBG, LRMK enhances both its HLA-DR and its TCR binding. If AE-37 binds its N-terminal foreign peptide (P-10 to P-2) to HLA-DR-PBG, R P-9 and V P-5 will point inside the TCR CDR3-α domain and will be able to activate T-cells specific for a foreign Ag (Table II, HLA-binding preferences).
The immunodominance of the AE-37 (LRMK-p776) depends on the healthy donor and the absence of εV. Essential proof of immunogenicity and immune memory is seen when cells rapidly respond to the immunogen without previous vaccination. The amount of IFN-γ produced by PBMCs activated by all AE peptides was quantified in 5 donors between 11/2003 and 7/2004. AE-37 was immunodominant in 3, AE-38 in 1, and G89 in 1. AE-37 activated 4 out of 5, AE-38 3 out of 5, AE-39, AE-47 and AE-48 each activated 2 out of 4, while G89 activated 3 out of 3 PBMC samples from distinct individuals. In absolute terms, AE-37 appears immunodominant (3/5= 60%). Surprisingly G89 and Ii-key-truncated G89, AE-38, were immunodominant. (1/4+1/4) (Table III). εV Appeared to decrease immunogenicity of p776. We do not know if LRMK and LRMK-εV enhanced or inhibited T-cell activation compared with free p776.
AE-37 is immunodominant in healthy donors.
The immunodominance of AE-37 and G89 depends on the BRC patient. We used AE-37 (immunodominant), AE-39 (εV-control for AE-37), AE-47 (negative control for GV of AE-39), and G89 (free peptide) to activate PBMCs from BRC patients between 1/2004 and 8/2004. LRMK-extended peptides, but not G89 or E75, induced death in 40-60% of PBMC at concentrations higher than 5 μg/ml (2500 nM). More cells died when AE-37/AE-47 were used together with E75. AE-37 and AE47 did not induce cell-death at 1 μg/ml (approximately 500 nM). We could not detect IFN-γ at priming with Ag unless we increased the amount of Ag to 10-25 μg/ml (5000-12500 nM). The death-effect required more starting cells for each experiment than we anticipated.
At priming, AE peptides and G89 induced 2- to 4-fold increases in IFN-γ-levels compared with cytokines IL-12+IL-2 only (Table IV). A similar increase was observed by ELISPOT, with few exceptions (Table IV, Murray et al., submitted). AE-37 was uniquely immunodominant in PBMC from 3 out of 11 patients, G89 in 3 out of 10 patients and AE-39 in 1 out of 11 patients. PBMCs from 2 patients responded both to G89 and AE-37, and both to G89 and AE-39. Patients' helper Ag preferences were similar to healthy donors' helper preferences (Tables III and V).
Patients with mixed disease, without metastases, preferentially responded to AE-37, while those with luminal cancer to G89. Surprisingly, each of the 3 patients with basal cancer had different responses. One responded to AE-37, AE-39 and AE-47, the second to AE-37 and G89, while the third only to AE-39 (Table V). We concluded that if the candidate helper AE-37 cannot activate help at priming it is better for the patient to be vaccinated with the helper Ag (G89, F7) which activates help at priming, than to attempt to elicit help at the third vaccination with AE-37, which is inactive in the particular patient for two vaccinations in a row. Our conclusion is supported by the original report on p776 which, induced responses after vaccination in 2 out of 4 patients (50%), but 0 out of 4 (0%), detectable responses before vaccination (3). However, our technology is more advanced and sensitive than non-quantitative methods used in the previous decade.
Relationship between the IFN-γ induction by AE peptides and HLA-DR phenotype of patient. The HLA-DR-type of 8 patients was identified. AE-37 is 4 amino acids longer than p776 and 7 amino acids longer than G89; it can bind HLA-DRB1* 0801 and HLA-DRB1* 0806 through its second minimum peptide, LRMKGVGSP, and HLA-DRB1* 0102 through its third peptide, MKGVGSPY (Table II). If the second and the third peptide are presented, T-cells will not recognize HER-2 peptides. We do not know how εV-containing peptides bind to HLA-DR.
Patient 2 (HER-2 Hi, DRB1*1, 13) and patient 3 (HER-2 Hi, DRB1*15 homozygous) responded to AE-37 and not to G89. Patient 5 (HER-2 Low, DRB1* 7, 13) responded to G89 and AE-39 but not to AE-37. HLA-DRB1* 07 is not preferred by the minimum HER-2 peptide; it probably binds each peptide with much lower affinity than DR101, 1307, 1501 and 1502 (Table II). Therefore the differences in response may be due to the presence of 2-peptide binding DR-molecules in patients 2 and 3 compared with one in patient 5.
IFN-γ responses to HER- 2 peptides by BRC patients.
Patient 4 (HER-2Hi, DRB1*04, homozygous), Patient 9(3) (HER-2Low, DRB1*01, 04), Patient 10(4) (HER-2Low, DRB*01 homozygous) and Patient 13(12) (HER-2Low DRB1*04, homozygous) responded to AE-37; AE-37, AE-39 and AE-47; AE-37 and G89; and AE-39 only, respectively. Results suggest that G89 and AE-37 bind only DR-1, while AE-39 can bind DR4. DR4 is not preferred by this HER-2 minimal peptide. Thus, either AE-39 binds DR4 (and DR7) due to LRMK-εV, or Patient 4 has mutations in the DR-β-chain. Studies with characterized HLA-DR (31) are needed to address this question.
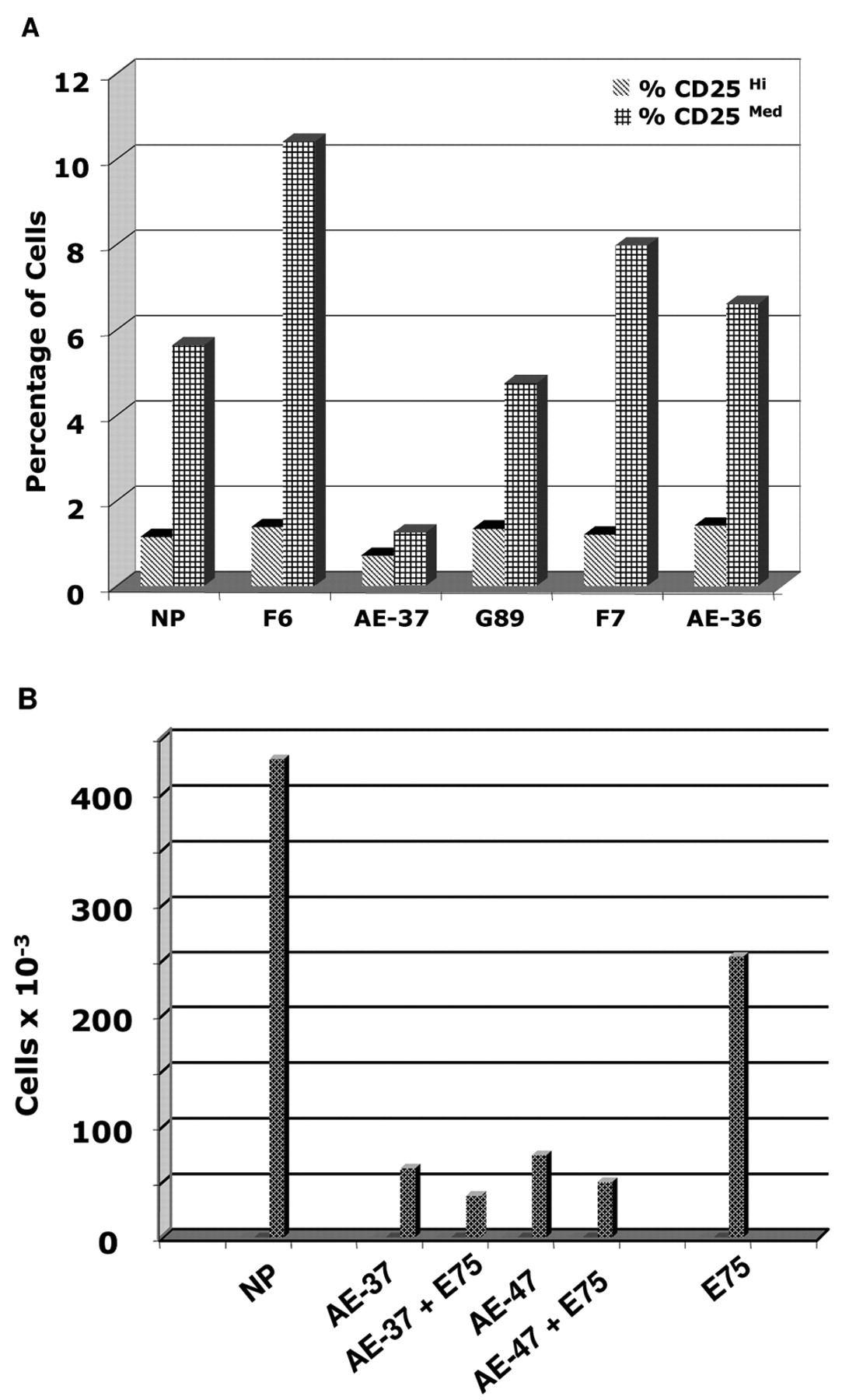
The magnitude of help for expansion of E75-TCR+ CD8+ cells inversely correlated with concentration of AE-37. Proof of help is the helper Ag to augment responses by, or synergize with, the helped. To identify whether AE-37 synergized with E75, we quantified the amount of IFN-γ produced at activation by all AE peptides, alone, and together with E75. PBMCs from patient 4 did not respond to AE peptides at priming in the absence of IL-12. In the presence of IL-12, PBMCs responded more strongly to AE-36 and AE-47 (>6-fold increase), weakly to AE-48 (2- to 3-fold increase), AE-37 and to G89, and inhibited IFN-γ production (AE-38 and AE-39). Only AE-47 synergized with E75, as demonstrated by the increase in IFN-γ levels above the sum (Σ) of the amount of IFN-γ produced in response to each antigen alone. Possibly the other peptides induced apoptosis of responders at recall as in PBMCs from donor 1. This finding shows a complex relation between “potential help” measured as IFN-γ produced at priming and “qualitative help” when both CD8+ and CD4+ cells were activated (Figure 3A).
Split immunodominance of AE-37 and G89 in mixed and luminal BRC patients.
To define the help for CTL expansion, in November to December, 2004 and the January to February 2005, Dr. Efferson found that AE-37 helped expansion of E75-TCR+ cells in PBMC from patient 3 (data not shown). In May 2005, Dr. Tsuda studied in parallel the help by AE-37 and AE-47 (negative control in IFN-γ induction, Table IV) for expansion of E75-TCR+ cells. In PBMCs from patient 4, restimulation with AE-37 (p776) and AE-47 (F7) reduced the number of live E75-TCR+ cells compared with cytokine-only cultured cells. Fewer AE-47- and AE-37-activated E75-TCR+ cells survived than E75-activated cells; more AE-47+E75-activated E75-TCR+ cells survived than AE-37+E75-activated E75-TCR+ cells (Figure 4).
The MFI of E75-TCR+ cells activated by AE-47+ E75 and AE-37+E75 were 2.3 and 4.1 times higher than that of cells activated by E75 plus cytokines or cytokines only. The MFI of CD8 was also two times higher. Therefore, AE-37 primarily, and AE-47 secondarily, alone, or together with E75 protected more cells with high density of E75-TCR+ and CD8 per cell than did E75 alone. Such cells can recognize smaller numbers of E75-HLA-A2 complexes on tumors (Figure 4). They had more perforin per cell than cells activated by other peptides (data not shown).
Dr. Matsueda repeated activation (April 2006), with 10 μg AE-37, AE-47 and E75 in patient 5. E75-TCR+ cells were not expanded by AE-37 or AE-47, but more E75-TCR+ cells survived when activated by AE-37 than by AE-47 (Figure 3B).
In Dr. Matsueda's next experiment in 5/2006, 1 and 5 μg of AE-37 and AE-47 helped expand similar numbers of E75-TCR+ CD8+ cells from PBMCs from patient 6 /healthy donor (blind sample). At 1 μg/ml AE-37 was a stronger helper than AE-47 because it expanded more E75-TCR Hi + Med CD8+ cells. At 5 μg/ml, AE-47 was a stronger helper than AE-37 for the same reason. Although the percentage of E75-TCR+ CD8+ cells was similar in both cultures, the total number of live cells was 2 times higher in cultures activated by AE-47 (F7) than by AE-37 (p776) (Figure 5).
In AE-37, LRMK is followed by GV. GV is absent from AE-47, which has εV instead. AE-37 helped more at priming E75-TCR+ cell expansion and survival, and at a lower concentration than AE-47. AE-47 was more effective than AE-37 at higher concentration. Stronger help for CTL function, by AE-47 (named peptide D) than by AE-37 (peptide F) and AE-39 (peptide B), was reported in a different model (32, 33). Therefore, the one amino acid shift to the left from AE-47 to AE-37 increased both “the help” and the toxicity of AE-37.
Inhibition of CD4+ CD25Hi cell expansion by AE-37. In 2003, Drs. Ioannides and Kawano found that AE-37, in donor 2, reduced the number of CD4+ CD25Hi cells compared with AE-48 (34). Drs. Ioannides, Efferson and Tsuda continued this study (2004-2005). We counted 4-fold fewer CD4+ CD25Hi cells in PBMCs from donor 5 after priming with AE-37 compared with priming with lower amounts of E75, F7, G89 and AE-36 (Figure 6A). In 2004-2005, Drs. Ioannides and Efferson investigated the role of changes at the C-terminus of F6, HER-2 (776-793) in inhibition of proliferation and expansion of CD4+ CD25Hi cells (35).

A, AE-47 synergize with E75 to enhance production of IFN-γ. Patient 4 PBMCs were activated with (1) cytokines only or with (2) E75, (3) G89, (4) AE-37, (5) AE-38, (6) AE-39, (7) AE-47, (8) E75+ G89, (9) E75+AE-37, (10) E75+AE-38, (11) E-75+AE-39 and (12) E75+AE-47. Cells were rested and re-activated with the same CTL and helper Ag (day 1 after recall), (day 2 after recall). Σ=(Sum of the amount of IFN-γ produced by E75 and each of G89, AE-37, AE-38, AE-39 and AE-47. Synergy was shown by AE-47, no effect by AE-37, and inhibition by G89, AE-38, AE-39. B, AE-37 protects cells with more E75-TCR and CD8-molecules per cell from death than AE-47. PBMCs from patient 5 were activated with cytokine only (NP) or with 10 μg each of AE-37, AE37+E75, AE47, AE47+E75, and E75. Columns show the total number of E75-TCR Hi, E75-TCR Med, E75-TCR Low, and E75-TCR cells.
We counted 4- and 3.1-fold fewer CD4+ CD25Hi cells in PBMCs from Patient 4 after priming with AE-37 and AE-47 than with the same amount of E75. E75+AE-37 reduced CD4+ CD25Hi cells by 6.7-fold, while E75+AE-47 reduced CD4+ CD25Hi cells 5.1-fold compared with E75 alone (Figure 6B). This effect was confirmed in one healthy donor and one patient (total 4 of 4). This effect is due to LRMK, since AE-36 and G89 did not reduce the number of CD4+ CD25Hi cells. However, overall elimination of cells by AE peptides raised questions of whether CD4+ CD25Hi cell elimination had additional contributing factors. Our findings were expanded by Dr. George Peoples' laboratory, as presented to AACR by Dr. von Hofe (36).

AE-37 and AE-47 helped E75-TCR+ CD8+ cell expansion. E75-TCR+ CD8+ cells activated by (A) cytokine only (NP), and (B) AE-37, (C) AE-37+E75, (D) AE-47, (E) AE-47+E75, and (F) E75. Total surviving cells (×10-6) were: NP=15.4, AE-37=2.8, AE-37+E75=1.3, AE-47=2.56, AE-47+E75=3.9, and E75=15.4. To obtain the number of cells listed in each column, the % positive cells in the upper right gate was calculated in the relation to the total number of live cells listed above. Cells = [% positive cells in the gate -> (% positive cells of total) × (total live cells)].
Discussion and Conclusions
Identification of a novel HLA-DR-binding site. The objective was to increase the number of Th1-differentiated helper cells by varying the length of the N-terminal sequence of the minimum HLA-DR-binding-peptide HER-2 (780-788). The minimum HER-2 peptide (780-788) is extended with 6 amino acids in p776. LRMK-εV added 5 amino acids to the N-terminus of p776. The longest extension of 11 (5+6) amino acids, in AE-39 was short of 14 amino acids needed by Clip (77-90) to position itself in the “CLIP-allosteric site”. Instead K P-7 “nailed” LRMK with a salt bridge to DRα (49-52), which constitutes the novel inside binding site.
Immunodominance of AE-37. Six out of 11 (54.5%) patients responded strongest to AE-37; 4 out of 11 (39.1%) of patients responded strongest to G89. More AE-37 and AE-47 was needed to detect IFN-γ production (25 μg/ml) than to help expansion of existent E75-TCR+ and de novo activation of E75-TCR+ cells (1-5 μg/ml). Individual responses of patients for help require selection of the optimal helper for elimination of cancer stem cells resistant to chemotherapy. Most BRC start as hormone sensitive and progress to being hormone resistant, owing to changes in progenitors of stem cells over time (37-39).
AE-37 reduces the number of CD4+ CD25Hi cells. This effect was observed in the context of massive death of all populations, which decreased by 2- to 5-fold the number of live cells compared with G89 and E75. The reason for the high numbers of dead cells at activation must be investigated; it may be the intrinsic toxicity described for Clip (77-82 and longer) at more than 5-10 μM together with induction of other cytokines.

Inverse correlation between AE-37 concentration and its help for E75-TCR+ CD8+ cell expansion. A-D, Cells from patient 6/donor 6 activated by E75 and AE-37. E-F, Cells activated by E75 and AE-47. The number of resulting live E75-TCR Hi, E75-TCR Med and E75-TCR Low cells was counted from the total number of live cells before staining. Note the similar increase in E75-TCR Hi, E75-TCR Med cells activated by 1 μg/ml AE-37 and 5 μg/ml of AE-47.
Unresolved questions. Are there one or two CD4+-responding populations which help the CD8+ cells?
Are effects of AE peptides due to their recognition by both CDR3 and CDR1 regions of the same T-cell? Contrary to processed antigenic peptides, SAG binds to MHC molecules outside of their PBG and interacts only with the V-β domains of TCR, resulting in the stimulation of up to 20% of the entire T-cell population. In this way, SAG initiates a systemic release of inflammatory cytokines, which, in most instances, is followed by responder death (40). Therefore AE-37 is not SAG.
Figure 6.AE-37 reduced the % of CD4+ CD25Hi+Med cells. PBMCs from donor 5 (A) and patient 4 (B) were activated with the peptides listed above. F6=F7 extended to C-terminus. PBMCs from patient 4 were activated with 25 μg/ml each of the NP, AE-37, AE-37+E75, AE-47, AE-47+E75 and E75. Total surviving cells (×10-6) were 5.4, 2.8, 1.3, 2.56, 3.9, and 15.4, respectively. Live cells were counted 6 days after activation. Cell death was not observed at 1 μg/ml.
Is help mediated by cytokines and can it bypass the need for re-activation of CTL by tumor antigen-specific Ag? It is unlikely that there is a wide-enough CDR3 region which accommodate R P-9, V P-5, Y P-1 and R P3 spanning 11-12 amino acids over 40 Å. TCRs contact a span of 25 Å between P-1 and P-8 (24). The number of E75-TCRHi + Med tumor-Ag-specific CD8+ cells increased in cultures activated only with AE-37. We propose that there are T-cells that recognize Y P-1 and R P3 in G89, AE-37 and AE-47. Other T-cells recognize R P-9 and V P-5 with low affinity, if the peptide P-10 to P-2 is in the PBG (Table I). At high AE-37 concentration, the other T-cells produce a burst of inflammatory cytokines and then die. These cytokines “feed and mature” few E75-TCRHi + Med cells.

Recommendations for future studies.
Define the qualitative help. Help for expansion and differentiation of tumor-specific CTL was found at an AE-37 concentration 10-20 times lower than the one used for IFN-γ induction. Screening before vaccination should include screening for help for differentiation of CTL with the optimal helper Ag for the patient.
Correctly position the LRMK-εV for qualitative help. Introduction of εV or movement of LRMK-εV changed the immunodominance. The Gly-amino (α) → εV -carboxyl (α) → εV- amino (δ) → Lys-carboxyl- (α) bonds either move LRMK 4 carbons to the left, or the 4 carbons bend and form a hairpin to accommodate an anchored K P-7 to DR. This question must be addressed.
Broaden the activation range of G89 and enhance its Th1 effect by creating mosaic tumor antigens. Since AE-37 is used to treat patients, there are plans to develop LRMK-based, Th1-helper antigen with a broader range. The main TCR contacts of self-peptides are P2 and P3, within the PBG; the secondary contacts are in P-1 (outside the PBG) and P5 (40, 41). These positions are identical to those of F7, G89 and p776 contacts, Y P-1 and R P3 for TCR (38).
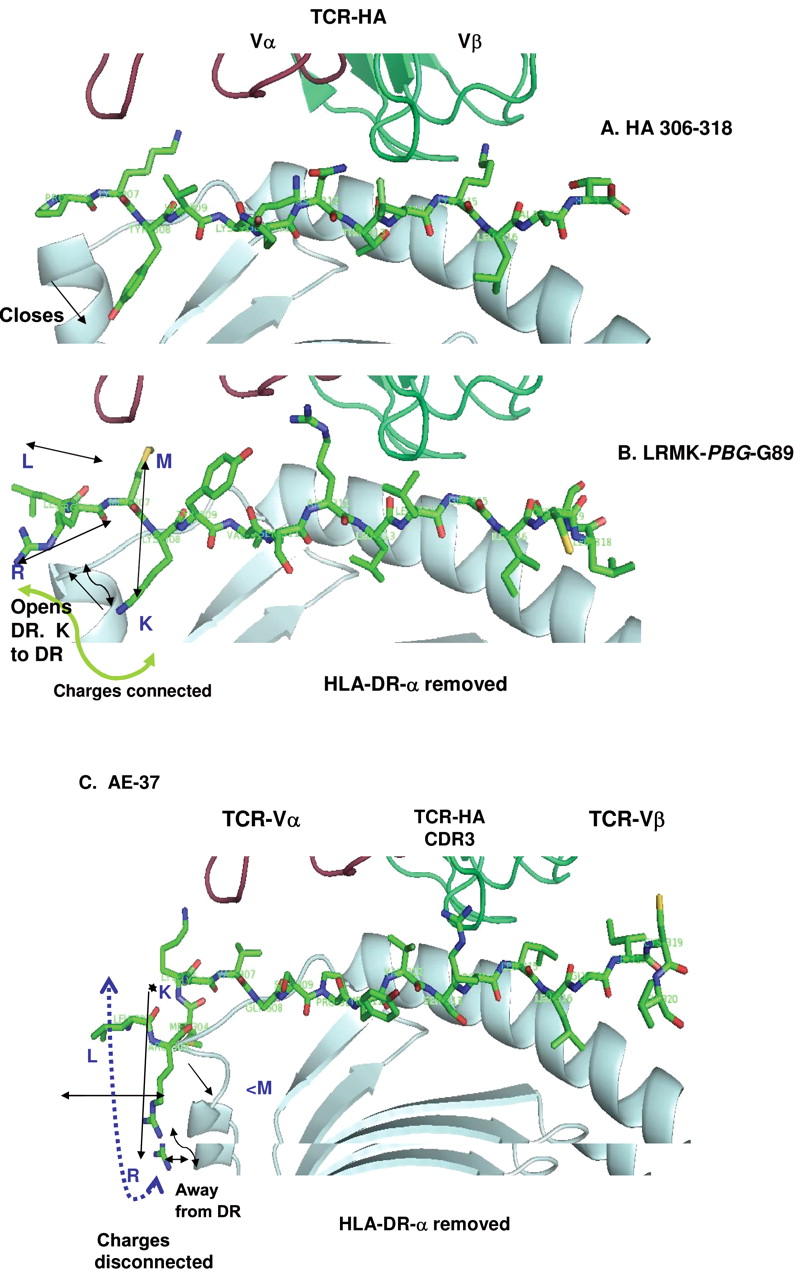
G89-T P10 can contact TCR-CDRβ2. A broader G89 range can be achieved with LRMK-YVSRLLGICLT (Figure 7). The positioning of the amino acid sequence P-3 to P3 is almost identical in G89 and influenza virus-HA1-peptide (306-318); G89= SP-YV-SR, HA = PK-YV-KQ suggesting that the peptide will cross-activate clones of both specificities. LR-MK-YV linked to P3-P10 of HER-2 (SRGICLT) should create mosaic HER-2 helper peptide that may more strongly activate more cells with diverse HLA-DR phenotypes. The HA1-peptide is a Th0 cell activator, with the environment turning it to Th2 helper for production of antibodies to influenza virus. HA1 peptide (306-318) is transformed in a Th1-antigen when the side chain of Val P1 is shortened to zero or 1 methylene groups (41). Maybe this is the direction to follow.
The mosaic peptide brought LRMK closer to CDR3 (Figure 7B). The strophynx turned R P-9 down and K P-8 up, but is less extended outside DR1 than AE-37 (Figure 7B, C). CDR3 can recognize the N-terminus of the minimum peptide in PBG (and by extension the self-antigens plus LRMK) (42, 43). Patients will benefit from more drugs with an extended range with finely tuned help. L P-10 and R P-9 in AE-37 are farther from CDR3α and more likely contact CDR1α.

Predicted structure of LRMK-YVSRLLGICLT peptide (LRMK-(PBG)-G89), using the PDB-1FYT file for influenza HA1 peptide-linked to TCRVβ through octa-Gly (24). A, HA1 (306-318). The model of HA1 (306-318) was made with the same file. Changes of V P1 to G or A do not change the positions of the neighboring side chains. B, LRMK-PDG-G89. K P-2 points down inside the third helix, moves closer to the PBG. M P-3 side-chain points up; R P-2 side chain is outside the helices of DR-β-chain. Possibly LRMK-PBG-G89 contacts TCRVα2 via methionine. The positive charges are connected in one area. Double arrows show orientation of the LRMK. Single arrow pointing upwards show the change in DR-N-terminus (open conformation) compared with HA (306-318) (downward arrow). C, AE-37. R P-9 is close to the outside of the first DR-β-helix; KP-7 points upward. Leu is intercalated between R and K, disconnecting the charged field. LRMK functions as rotating bi-strophynx/Sanchaji.
A newly described shallow pocket on the edge of the MHC peptide binding site interacts with peptide P10 side-chains, and common MHC polymorphisms in this region alter the peptide side-chain specificity. Our findings with G89 suggest that P10 is important in HER-2 peptide selectivity (44, 45). Therefore the choice is between quantifying the qualitative help of AE-37, AE-47 and G89 of the HLA-typed patient to find the optimal helper for the particular patient and their type of BRC and attempting to broaden the range of G89 by mosaic peptides for all patients.
Reduce the SAG-like effects of LRMK. It is possible that L P-10 and R P-9 create additional TCR contacts to CDRVα1 and activate cells non specifically. If toxicity of AE-37 is due to other reasons information must be provided.
Antigens such as AE-37, AE-47, AE-39 and LRMK-εV-G89 are novel. LRMK, and LRMK-Ava cannot be fused with all and any HLA-DR-bound peptides of any length and become Th1-helpers. We suggest how to extend the “outside chains”. Our findings should be of interest to the BRC patients now treated with the “protected” - AE-37, LRMKGVGSPY VSRLLGICL, in USA and Europe, and patients with other types of cancer.
Financial interest: Ownership of the tumor antigens used in this study and of the results of this research.
P776 (3), discovered by Drs. Disis and Cheever, and other peptides, p793, p781, p788, reported earlier belong to Fred Hutchinson Cancer Center, Seattle (46). AE-36, AE-37, AE-39 and AE47 contain the sequences LRMK and LRMK-Ava, respectively. LRMK and LRMK-Ava are owned by University of Massachusetts and AE. Inc/Generex (47). E75 and its analogs are joint patents MDACC and Henry Jackson Fundation (48, 49). F7, property of University of Texas System Board of Regents, was discovered by Drs. Ioannides, Bryan Fisk and Maria Ioannides. F7 is the first CD4 cell recognized minimum antigen to activate T-cells in donors and patients (4, 50). F7 was followed by G89 (51, 52). AE-47 contains the F7 (13 amino acid long). AE-47 (without this name) but linked to LRMK +/- Ava was submitted as US Patent in 9/24/2002. The patent was assigned to AE. Inc on 3/25/2004 (53). G89, property of University of Texas System Board of Regents (51), was discovered by Dr. Ioannides. G89 with IL-12 as co-factor was submitted to MDACC as Invention Disclosure Report, (MDA-0043.3/2001) and to AE. Inc for evaluation for submission of a joint patent.
AE. Inc. asked Dr. Ioannides in 5/2000 for help (5). SRAs were made between AE. Inc and MDACC (11/8/2000), to study Ii-key-G89-HER-2 (777-789) (54). Because initial results of Dr. Ioannides were promising, a second SRA, LS2003-00009873SP (07/15/2003), was executed. This SRA: (a) defined allocation of intellectual property to be created and obligations of parties in a Research Program; (b) agreed to study Ii-key-HER-2, 777-789, and (c) confirmed AE. Inc. duty not to commercialize the findings under it (55-57). The agreed Ii-key G89 (777-789) was substituted in 11/2003 with 6 AE-peptides (Table I). After receiving Dr. Ioannides' results, Dr. Humphreys (12/2003) requested from Drs. Murray and Ioannides to change work-scope from MDACC-owned G89 to University of Washington-owned p776 +LRMK. The amended contract with provisions for equity compensation of Dr. Ioannides and of his laboratory for the study of University of Washington-owned p776 was not made yet.
The original findings of Dr. Ioannides' laboratory on AE-peptides were reported and confirmed with AE peptides named A, B, C, D, E, and F (32). We do not know if AE-peptides and A-F peptides are identical (31, 32). Consequently, although Dr. Ioannides received no remuneration, he and the members of his laboratory have joint financial interest with MDACC in the income from commercialization of their findings, starting 1994 and until today, by others.
Acknowledgements
We thank the 26 breast cancer patients who donated their blood for these studies. We thank Dr. Juergen Hammer (Roche) for the TEPITOPE Program. We also thank Dr. John Judge for making this paper possible through his expert study of complex SRA and Requests for Work-changes in scope to us from AE. Inc. in 12/2003. We thank Dr. Alexander Papandreou for selection of the name bi-strophynx for the structure of LRMK.
This study was supported by funds from the M.D. Anderson Cancer Center (CGI), Graduate School of Biomedical Sciences (YL), DOD-grant-01-1-0299 (SI, NT, CGI), Patent Licensing Funds to CGI, the SRA-LS2003-00009873SP (07/15/2003) from Antigen Express, Inc. (KK, NT), Keck Foundation (CGI), NIH-MDACC Core Grant CA-16660 to Peptide Synthesis Laboratory, Eustathios and Euphrosina Maroulis Memorial Fund, The European Union “Socrates” and “Jean Monet” Programs, funds from Maria G., and Fotini G. Ioannides (Athens, Greece) to CGI, Drs. John Zavitsanos and Paul Botros and The Department of Defense of USA, US Military Cancer Institute (GEP).
Footnotes
-
↵* These authors contributed equally to this work.
- Received June 19, 2008.
- Revision received August 12, 2008.
- Accepted September 22, 2008.
- Copyright© 2009 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}