


Abstract
Background: Curcumin, an active ingredient of turmeric with no discernable toxicity, inhibits the growth of transformed cells and the development and progression of colon carcinogenesis in experimental animals. Recent data from one of our laboratories demonstrated that a crude skin extract or a purified crystalline compound (Bufo melanostictus-antineoplastic factor 1, BM-ANF1) from Indian common toad (Bufo melanostictus, Schneider) skin inhibits the growth of human leukemic cells. The present investigation was undertaken to determine whether combining BM-ANF1 with curcumin would be a better therapeutic strategy for colon cancer. Materials and Methods: Colon cancer HCT-116 cells were used. Changes in growth, apoptosis, growth factor receptor signaling and events of the cell cycle were analyzed. Results: Curcumin together with BM-ANF1 produced a greater inhibition of HCT-116 cells growth than either agent alone, attributable to the inhibition of proliferation and stimulation of apoptosis, as evidenced by suppression of proliferating cell nuclear antigen (PCNA) expression, cell cycle arrest at the G2/M-phase and caspase-3 activation. There was also a marked reduction of cyclin-dependent kinase (CDK)2, CDK4 and cyclin B expression and up-regulation of CDK inhibitors (p21, p27) and p53, accompanied by attenuation of Akt signaling and nuclear factor-kappa B (NF-κB) activation. Conclusion: BM-ANF1 in combination with curcumin causes a marked inhibition of growth of colon cancer cells and could be an effective therapeutic strategy for colon cancer.
Despite recent advances in medicine, mortality from colorectal cancer, the third most common malignancy among men and women, remains unacceptably high. This has led to a surge in research for a more effective therapeutic strategy for colorectal cancer. Indeed, in view of the fact that colorectal cancer is associated with increased expression and activation of several growth factor receptors, including epidermal growth factor (EGF)-receptor (EGFR) and its family members, a number of drugs such as cetuximab, trastuzumab, gefitinib and erlotinib that target either EGFR or human epidermal growth factor receptor (HER)-2 have been developed, but with limited success (1-3). Recent data have also implicated the insulin-like growth factor (IGF)/IGF-1 receptor (IGFR) system in the development and progression of colorectal cancer (4-6). Since multiple signal pathways are activated during the development and progression of malignancy, combination therapy that could target different intracellular events may prove to be more beneficial than monotherapy.
Curcumin (diferuloylmethane), the major active ingredient of turmeric (Curcuma longa) with no discernable toxicity, has been shown to inhibit the growth of transformed cells (7, 8) and colon carcinogenesis at the initiation, promotion and progression stages in carcinogen-induced rodent models (9-11). Curcumin prevented the development of intestinal adenomas in Min+/- mice, a model of human familial adenomatous polyposis (12). In a Phase I clinical trial, curcumin was shown to inhibit the growth of a variety of tumors (13). We have recently demonstrated that curcumin together with ERRP (EGFR-related protein), which we have shown to be a pan-erbB inhibitor (14), caused a greater inhibition of growth of colon cancer cells in vitro than either agent alone (15). This was partly attributed to the attenuation of signaling induced by EGFRs and IGF-1R leading to inhibition of nuclear factor-kappa B (NF-κB) (15). Additionally, we have demonstrated that curcumin in combination with either 5-fluorouracil or FOLFOX (5-fluorouracil and oxaliplatin) caused a greater inhibition of growth of colon cancer HCT-116 and HT-29 (p53-positive or p53 mutant) cells than that caused by either agent/regimen alone (16). This inhibition was also attributed to a reduction in expression and activation of EGFRs and IGF-1R and their signaling pathways (16). These studies raise the possibility of investigating the therapeutic strategy of combining curcumin with other antineoplastic agent(s) for colorectal cancer.
It is known that amphibians such as the frog and toad possess different bioactive substances in their skin (17). In Chinese traditional medicine, amphibian skin extract has been used for the treatment of different diseases (18, 19). Chan'Sue, prepared from Chinese toad (Bufo gargrizans) skin extract has been used in the treatment of diseases, including cancer (20-22). Bufalin (a bufadienolide), one of the active components of Chan'Sue, induced differentiation and apoptosis in human leukemia HL60 cells (23, 24).
Recently, Giri et al. reported that the skin extract from the Indian common toad (Bufo melanostictus Schneider) inhibited the growth of leukemic U937 and K562 cells (25). Subsequent studies utilizing the purified (crystalline) product of the skin extract (referred to as BM-ANF1 [Bufo melanostictus-antineoplastic factor 1]), further revealed the growth inhibitory properties of this compound in human leukemic (U937 and K562) and hepatoma (HepG2) cells, as evidenced by the suppression of proliferation and induction of apoptosis, accompanied by cell cycle arrest and up-regulation of p53, p21cip1 and p27kip1 (26). The current in vitro investigation was undertaken to determine whether combining BM-ANF1 with curcumin would be an effective therapeutic strategy for colon cancer.
Materials and Methods
Chemicals. Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and antibiotic/antimycotic were obtained from GIBCO BRL (Bethesda, MD, USA). Curcumin was purchased from Sigma Chemical Co. (St. Louis, MO, USA). Antibodies to caspase-3, caspase-9, Akt, anti-p-Akt (Ser473), anti-Bad and anti-p-Bad were purchased from Cell Signaling Technologies (Beverley, MA, USA). Antibodies to cyclin-dependent kinase (CDK)4, CDK2, p53, cyclin B, p21 and p27 were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). β-Actin antibody was purchased from Sigma Chemical Company. NF-κB binding consensus oligonucleotide was procured from Li-COR Inc, (Lincoln, NE, USA). Crystalline BM-ANF1 was prepared by one of the authors (B.G.) in the Laboratory of Toxinology and Experimental Pharmacodynamics, University of Calcutta, Kolkata, India.
Cell line and cell culture. Human colon cancer HCT-116 cells were obtained from American Type Culture Collection (ATCC, Rockville, MD, USA). The cells were maintained in tissue culture flasks in a humidified incubator at 37°C in an atmosphere of 95% air and 5% CO2. The medium (DMEM) was supplemented with 10% FBS and 1% antibiotic/antimycotic. The medium was changed three times a week and the cells were passaged using trypsin/ethylenediamine-tetracetic acid (EDTA).
Growth inhibition assay. The inhibition of cell growth in response to curcumin and/or BM-ANF1 was assessed by 3-(4,5-dimethyl-thiazol-2yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay as described previously (15, 16). Briefly, the cells were dispersed by trypsin-EDTA treatment and 2.5×104 cells/ml, resuspended in DMEM containing 10% of FBS, were seeded into 96-well culture plates with six replicates. After 24 h, the medium was replaced with one that contained 2.5% of FBS to minimize the contribution of serum-derived growth factors and the incubation was continued for another 48 h in the absence (control) or presence of curcumin or BM-ANF1, or a combination of both as described in the figure captions. At the end of the 48 h incubation period, the reaction was terminated by adding 20 μl of 5 mg/ml stock MTT to each well. The reaction was allowed to proceed for 3 h at 37°C. The culture medium was then removed. The formazan crystals were then dissolved by adding 0.1 ml of dimethyl sulfoxide (DMSO). The intensity of the color developed, reflecting the number of live cell, was measured at a wavelength of 570 nm. All the values were compared to the corresponding controls. All the assays were performed with six replicates.
Assessment of apoptosis. Approximately 1×105 cells/well were plated in DMEM/10% FBS. After 24 h, the medium was changed to contain 2.5% FBS and subsequent treatment was as described above for growth inhibition. At the end of the incubation period, the cells were lysed, and the levels of apoptosis were determined using a Cell Death Detection ELISAPLUS kit from Roche Diagnostics GmbH (Penzberg, Germany), which measured the cytoplasmic histone-associated-DNA-fragments (mono- and oligonucleosomes). The levels of apoptosis were further validated by measuring the relative abundance of the activated form of caspase-3 by Western blotting as described below.
Western blot. Western blot analysis was performed essentially according to our standard protocol (27). Briefly, the cells were solubilized in lysis buffer. Following clarification at 10,000g for 15 min, the supernatant was used for Western blot analysis. In all the analyses, the protein concentration, determined by Bio-Rad protein assay kit (Bio-Rad, Hercules, CA, USA), was standardized among the samples. Aliquots of cell lysates containing 50 μg of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Following electrophoresis, the proteins were transferred electrophoretically onto nitrocellulose membranes (Osmonics, Gloucester, MA, USA). The membranes were incubated for 1 h at room temperature with blocking buffer, TBS-T (20 mM Tris, pH 7.6, 100 nM NaCl, 0.1% Tween-20) and 5% nonfat dry milk with gentle agitation. After washing the membranes with TBS-T, they were incubated overnight at 4°C in TBS-T buffer containing 2.5% milk and with one of the antibodies of the following (1:1,000 dilution): CDK2, CDK4, cyclin-B, p21cip1, p27kip1, p53, p-Akt, p-Bad, PCNA, caspase-3, caspase-9. The membranes were washed three times with TBS-T, and subsequently incubated with appropriate secondary antibodies (1:5,000 dilution) in TBS-T containing 2.5% milk for 2 h at room temperature with gentle shaking in a rocker. The membranes were washed again with TBS-T and the protein bands were visualized by enhanced chemiluminescence (ECL) detection system (Amersham Bioscience, Piscataway, NJ, USA). The membranes containing the electrophoresed proteins were exposed to X-Omat film, and the signals were quantified by densitometry using an ImageQuant image analysis system (Storm Optical Scanner; Molecular Dynamics, Sunnyvale, CA, USA). The membranes were stripped (2 × for 15 min at 55°C) in stripping buffer containing 100 mM 2-mercaptoethanol, 2% sodium dodecyl sulfate and 62.5 mM Tris-HCl pH 6.7. The membranes were then reprobed for the levels of β-actin which was used as a loading control. All the Western blots were performed at least three times for each experiment.

Effect of curcumin and BM-ANF1 on (A) growth of colon cancer HCT-116 cells, as determined by MTT assay, *p<0.01, +p<0.001, compared to curcumin or BM-ANF1 alone. Each value represents mean±SEM of 4 observations; (B) levels of PCNA (proliferating cell nuclear antigen) as determined by Western blot analysis.
Cell cycle analysis. Seeded HCT-116 cells were grown to about 50% confluency then treated with either curcumin (10 μM) and/or BM-ANF1 (20 μg/ml) and incubated for 2 days. Floating cells were collected by centrifugation. The attached cells, following treatment with trypsin, were washed with cold PBS and combined with the floating cells. The cells were fixed in 70% ethanol and stored at -20°C overnight. The cells were then incubated in the staining solution (10 μg/ml propidium iodide, 50 μg/ml RNAse, 0.1% Triton® X-100 and 0.1 mM EDTA) for at least 30 min at 4°C before being subjected to a BD Biosciences FAC Scan cytometer (BD Biosciences, San Jose, CA, USA) and analyzed by Modfit software.
Electrophoretic mobility shift assay (EMSA). Nuclear protein extracts were prepared according to the method described earlier by Banerjee et al. (28). Briefly, the cells were washed with cold PBS and suspended in 0.15 ml of lysis buffer (10 mM HEPES [pH 7.9], 10 mM KCl, 0.1 mM EDTA, 1 mM dithiothreitol [DTT], 1 mM phenylmethyl sulfonyl fluoride [PMSF], 2 μg/ml leupeptin, 2 μg/ml aprotinin, and 0.5 mg/ml benzamidine). The cells were allowed to swell on ice for 20 min and then 4.8 μl of 10 % Nonidet P-40 was added. The tubes were vigorously vortexed for a few seconds and centrifuged for two minutes in a microfuge. The nuclear pellet was resuspended in 30 μl of ice-cold nuclear extraction buffer (20 mM HEPES [pH 7.9], 0.4 M NaCl, 1 mM EDTA, 1 mM DTT, 0.5 mM PMSF, 2 μg/ml leupeptin, 2 μg/ml aprotinin, and 0.5 mg/ml benzamidine) and further incubated on ice with intermittent mixing. The tubes were then centrifuged for 5 min in a microfuge at 4°C, and the supernatant (nuclear protein extract) was collected in a cold eppendorf tube and stored at -70°C for later use. The protein content was measured by Bicinchoninic acid assay kit with bovine serum albumin BSA as the standard (Pierce Chemical Co., Rockford, IL). The EMSA was performed by incubating 8 μg of nuclear protein extract with IRDye™-700 labelled NF-κB oligonucleotide (Li-COR Inc, Lincoln, NE, USA). The incubation mixture included 25 mM DTT and 0.25% Tween 20 in a binding buffer. The DNA-protein complex formed was separated from free oligonucleotide on 8.0% native polyacrylamide gel using buffer containing 50 mM Tris, 200 mM glycine, pH 8.5 and 1 mM EDTA, and then visualized by an Odyssey Infrared Imaging System using Odyssey Software Release 1.1 (Li-COR Inc.).
Statistical analysis. Unless otherwise mentioned, data are expressed as mean±SEM. Where applicable, the results were analyzed either by Student's t-test or by ANOVA taking p<0.05 as the level of significance.
Results
Growth of HCT-116 cells. Following incubation with increasing doses of curcumin and BM-ANF1, for each combination there was a greater inhibition of cell growth (40-50% greater inhibition), as assessed by MTT assay than that caused by either agent alone, when compared with the controls (Figure 1A). These changes were accompanied by a concomitant reduction (65-70%) in the levels of PCNA (Figure 1B).
Apoptosis of HCT-116 cells. Following incubation with increasing doses of curcumin and BM-ANF1, for each combined treatment, there was a greater induction (30-40% more) of apoptosis than that caused by either agent alone, when compared with the control (Figure 2A).
Western immunoblot analysis revealed that whereas curcumin or BM-ANF-1 stimulated the levels of activated caspase-3 and -9 in the HCT-116 cells by 30-40% and 20-30%, respectively, curcumin together with BM-ANF-1 caused a much greater increase (60-70%) in the levels of caspase-3 and -9 when compared with the controls (Figure 2B).
Cell cycle distribution. The treatment with curcumin or BM-ANF1 resulted in the accumulation of cells at the G2/M-phase suggesting a G2 to M block in the cell cycle, but the number of cells arrested at the G2/M phase by combined curcumin and BM-ANF1 treatment was markedly greater (55.3% accumulation, i.e. about 25-30% more) than that caused by either agent alone (Table I).
Both curcumin and BM-ANF-1, individually reduced the levels of CDK2 and CDK4 by 30-60%; however together they caused a marked 85-90% reduction in CDK2 and CDK4, when compared with the corresponding controls (Figure 3A). As expected, the magnitude of reduction in CDK2 expression caused by curcumin, BM-ANF-1, or the combination was accompanied by parallel changes in cyclin B levels (Figure 3A). The levels of p21Waf1/Cip1 and p27kip1 in the HCT-116 cells were found to be markedly higher (2-3 fold) following 48 h incubation with the combination of curcumin and BM-ANF-1 than those noted with curcumin or BM-ANF1 alone (Figure 3B). These changes were associated with a rise in the levels of p53 but in this case BM-ANF1 produced a greater p53 induction than did curcumin (Figure 3B).

Effect of curcumin and BM-ANF1 on (A) apoptosis as determined by histone-associated-DNA-fragmention assay of colon cancer HCT-116 cells (*p<0.01, +p<0.001, compared to curcumin or BM-ANF1 alone). Each value represents mean±SEM of 4 observations. (B) Levels of activated forms of caspase-3 and 9 as determined by Western-blot analysis.
Akt phosphorylation. As shown in Figure 4, exposure to 10 μM curcumin produced a marginal reduction in the level of phosphorylated forms of Akt and Bad in the HCT 116 cells. BM-ANF1 on the other hand, produced a significant reduction in the level of the phosphorylated forms of Akt and Bad, compared to the untreated controls. Additionally, the combination of curcumin and BM-ANF1 produced a much greater reduction of phosphorylation of both Akt and Bad than that caused by either agent alone. The non-phosphorylated forms of Akt and Bad were not affected by any of the treatments (Figure 4).
DNA binding activity of NF-κB. The EMSA showed that whereas curcumin or BM-ANF1 reduced the DNA-binding activity of NF-κB in the HCT-116 cells by 20%, the combination of curcumin and BM-ANF1 caused approximately 60% inhibition when compared with the controls (Figure 5).
The effect of curcumin and BM-ANF1 on HCT-116 cell cycle distribution.
Discussion
The current data demonstrated that curcumin acted synergistically with the purified amphibian skin extract-derived secretory chemical BM-ANF1 to inhibit the growth of colon cancer cells. The results were agreement with our earlier studies using curcumin with ERRP (15) and with FOLFOX (16). Taken together, the results suggest that curcumin together with either conventional colon cancer chemotherapeutics or BM-ANF1 causes a greater inhibition of growth of colon cancer cells than either agent/regimen alone.
The fact that the magnitude of reduction of growth of colon cancer cells by the combination therapy was associated with a concomitant decrease in the levels of PCNA suggested that the growth reduction was partly due to the inhibition of proliferation. PCNA is an auxiliary protein for DNA polymerase δ that is expressed during the S-phase of the cell cycle and its levels have been shown to correlate positively with proliferation (29-31).
The suppression of apoptosis is one of the hallmarks of tumor formation. The combination therapy also caused a marked induction of apoptosis, as evidenced by increased DNA-fragmentation and supported by a parallel rise in cleaved caspase-3 and caspase-9 levels in the HCT-116 cells. The latter also suggested the involvement of the mitochondrial pathway in inducing apoptosis by curcumin and/or BM-ANF1 since caspase-9 is triggered by mitochondrial damage. We also reported that curcumin stimulated caspase-3 activity (15), an effector caspase that is downstream of the initiator caspase-9 which, in turn, regulates intrinsic apoptosis signaling pathways. Taken together, the results suggested that the comparatively greater stimulation of apoptosis of HCT-116 cells in response to the combination of curcumin and BM-ANF1 compared to that caused by either agent alone involved, in part, the caspase-9-dependent intrinsic apoptosis signaling pathway.

Effect of curcumin and BM-ANF1 on (A) the levels of CDK2, CDK4 and cyclin B and (B) the levels of p21, p27 and p53 in colon cancer HCT-116 cells, as determined by Western blot analysis.
The progression of mammalian cells through different cell cycle phases is a highly regulated process which is largely controlled by cyclins, the regulatory units of CDKs (32). For example, cyclin B is the primary regulator of CDK2. One of the events preceding apoptosis is cell cycle arrest. The observation that HCT-116 cells showed a greater G2/M arrest in response to the combination therapy than that caused by either agent alone suggested that apoptosis was triggered through G2/M arrest. At least for curcumin, this is in complete agreement with what has been reported by others in colon cancer cells (7, 33-36). The induction of apoptosis was further supported by the significant reduction in CDK2, CDK4 and cyclin B. CDKs are regulated by a class of CDK inhibitors (CDKIs), of which, p21Waf1/Cip1 (p21) and p27kip1 (p27) have been studied extensively and are considered to be universal inhibitors of different cyclin-CDK complexes (37). The fact that the expression of both p21 and p27 was markedly increased in response to the combination therapy also supports the contention that CDKs are intricately involved in cell cycle arrest. Although a major effect of p21Waf1/Cip1 is considered to be exerted during the G1 phase of the cell cycle, p21Waf1/Cip1 gene knock-out studies have suggested its involvement in the G2/M checkpoint as well (38). The induction of p21Waf1/Cip1 was also implicated in a G2/M arrest in both p53-proficient (39) and p53-deficient (40, 41) cancer cells. Clearly, p21Waf1/Cip1 has an effect on G2-M regulation because the introduction of nonfunctional p21Waf1/Cip1 or a p21Waf1/Cip1 antisense oligonucleotide diminished the G2-M arrest phenotype in various cancer cell settings (40, 41). The regulation of p21Waf1/Cip1 is largely dependent on the presence of functional p53, a transcriptional regulator that mediates cell cycle arrest following DNA damage (42). p53 plays a pivotal role in arresting cells during cell cycle progression whenever it is required. The fact that levels of both p21Waf1/Cip1 and p53 were increased in response to combination therapy, suggested a p53-dependent mechanism for p21Waf1/Cip1 regulation.

Effect of curcumin and BM-ANF1 on the levels of phosphorylated forms of Akt and Bad in colon cancer HCT-116 cells, as determined by Western blot analysis.
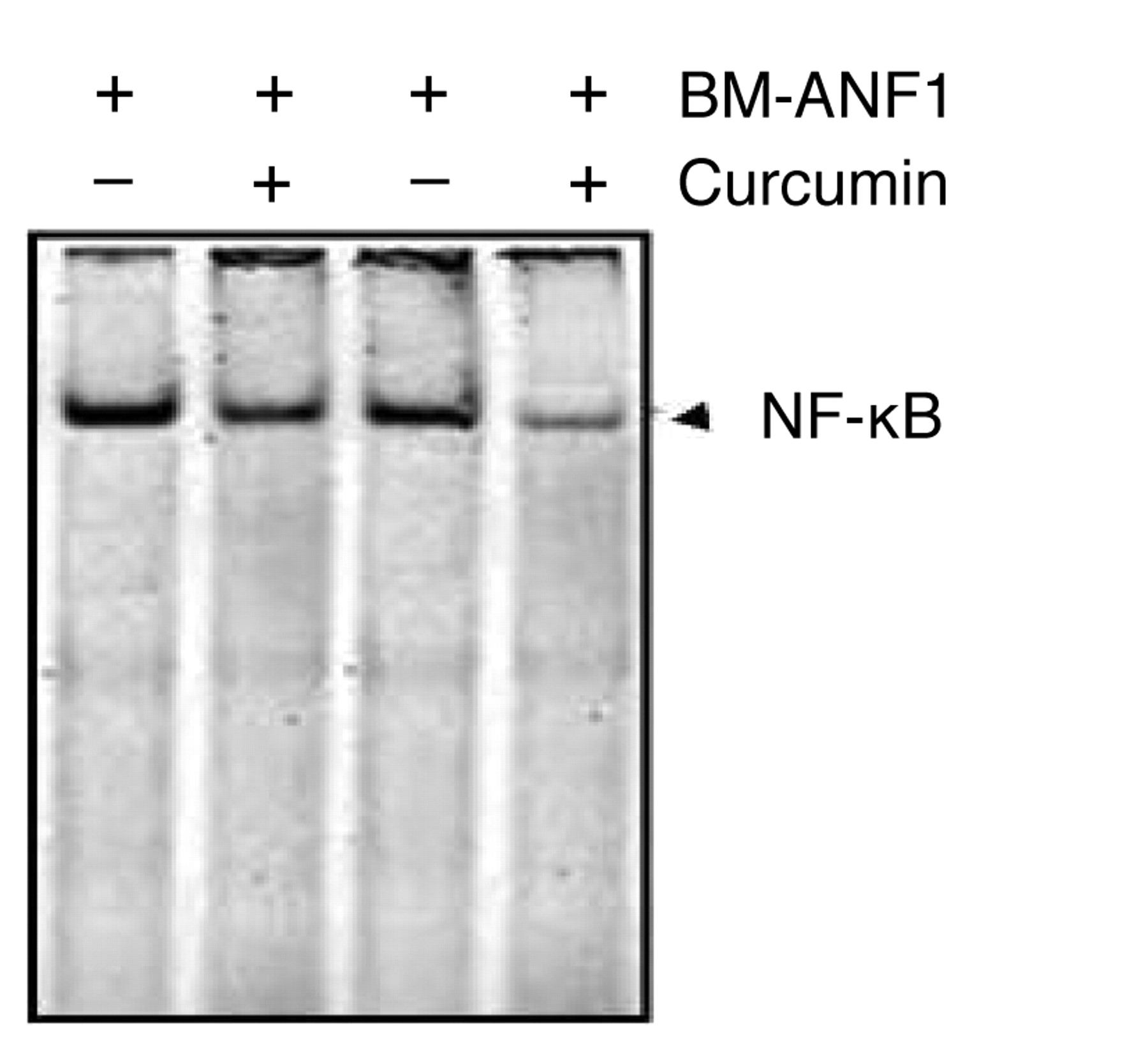
EMSA of the effect of curcumin and BM-ANF1 on the DNA-binding activity of NF-κB in colon cancer HCT-116 cells.
The combination therapy-induced apoptosis in the HCT-116 cells could be attributed to the attenuation of Akt signaling, as evidenced by a greater reduction in the levels of the phosphorylated (activated) form of Akt and its downstream effector protein, Bad. Akt phosphorylates Bad at Ser136 to promote cell survival (43). Non-phosphorylated Bad is known to promote apoptosis by binding to and neutralizing anti-apoptotic Bcl-2 proteins, whereas phosphorylated Bad is sequestered by 14-3-3 preventing Bad from blocking the anti-apoptotic action of Bcl-2 (44). Akt mediated stimulation of cell survival is transduced, in part, by activation of NF-κB (45, 46), which induces the expression of pro-survival genes including Bcl2 (47). Several studies, including our own, have demonstrated that curcumin-mediated inhibition of growth of certain epithelial cancer cells, including colon cancer, is associated with decreased activation of NF-κB (15, 48). The current observation of a greater reduction in DNA-binding activity of NF-κB in colon cancer HCT-116 cells in response to the combination treatment of curcumin and BM-ANF1 than that noted with either agent alone suggested the involvement of NF-κB in inhibition of growth in response to the combination therapy.
In conclusion, curcumin together with BM-ANF1 caused a substantially greater inhibition of cell growth than that achieved by either agent alone. This was associated with reduced proliferation and increased apoptosis, as indicated by the activation of caspase-3 and -9 and G2/M cell cycle arrest, which could be attributed to down-regulation of CDK2 and CDK4 and upregulation of CDKIs (p21 and p27). These events appear to be p53 dependent and regulated via Akt-NF-κB signaling. The combination of curcumin and BM-ANF1 could be a potential therapeutic regimen for colon cancer.
Acknowledgements
Author Biplab Giri and his mentor Professor Antony Gomes are grateful to the Society for Experimental Biology and Medicine, USA for providing the ‘Young Scientist Mentoring Award 2007’ to Biplab Giri to carry out a part of the work, presented in this manuscript in the laboratory of Professor Adhip P. N. Majumdar, Department of Internal Medicine, Wayne State University, Detroit, Michigan, USA. A part of the work was supported by grants to Dr. Majumdar from the Department of Veterans Affairs (VA Merit Review).
Footnotes
- Received July 10, 2008.
- Revision received September 1, 2008.
- Accepted October 29, 2008.
- Copyright© 2009 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.