


Abstract
Background: One of the most prevalent causes of cancer fatalities is hepatocellular carcinoma (HCC), which has been linked to metabolic syndrome. Circulating levels of the saturated fatty acid palmitate are elevated in metabolic syndrome and lead to cellular stress. Materials and Methods: Using enzyme-linked immunosorbent assay, flow cytometry, and migration assays, we characterized the response of rat hepatoma cells to palmitate treatment. Results: We detected a 60% increase in secretion of C-X-C motif ligand 1 (CXCL1) which was dose-dependent and coincided with apoptosis. We measured expression of C-X-C motif chemokine receptor 2 (CXCR2) and observed a 4.5-fold increase on apoptotic hepatoma cells. Furthermore, we assayed migration of hepatoma cells and saw a 2-fold increase in the number of migrating cells towards CXCL1. Conclusion: These findings suggest that HCC cells secrete CXCL1 in response to metabolic syndrome signals and may promote the progression of cancer through apoptosis recovery or metastasis.
Hepatocellular carcinoma (HCC) accounts for up to 85% of total liver cancer cases (1), and, due to an incomplete understanding of its epidemiology and very poor prognosis, is the second leading cause of cancer mortality worldwide (2). The majority of HCC cases develop from hepatitis C and B viral infection, followed by excessive alcohol intake; however, the most rapidly increasing cause of HCC is due to the epidemic of metabolic syndrome often in the form of type 2 diabetes, obesity, and non-alcoholic fatty liver disease. As the symptoms of HCC are commonly not apparent until an advanced stage of disease, the only method of obtaining an early and treatable initial diagnosis is surveillance. Ultrasonography of the liver and detection of serum alpha-fetoprotein levels every 6-12 months are the current recommendations for monitoring high-risk patients, such as those with metabolic syndrome (3). With obesity affecting 36% of U.S. adults (4) and ongoing concerns of patient compliance, it is inevitable that a developing HCC tumor is continually exposed to the lipotoxic and inflammatory cytokine milieu present in metabolic syndrome.
Elevated serum levels of free fatty acids (FFAs) are associated with obesity, caused by increased lipolysis in expanding adipose tissue (5), and a correlation exists between obesity and higher serum level of the saturated fatty acid palmitate (6). As non-adipose tissues are exposed to these excessive FFA levels, uptake may result in β-oxidation, synthesis of membrane phospholipids, and triglyceride synthesis and storage in lipid droplets (steatosis). If influx exceeds usage, its accumulation may lead to intracellular metabolites such as diacylglycerol, lysophosphatidylcholine, and ceramides to dysregulate signaling events downstream of other cell-surface receptors and cause lipotoxicity (7, 8). When faced with palmitate, HCC differs from hepatocytes in its response, since it is well known that cancer cells have departed from normal cellular physiology. A metabolic comparison of primary rat hepatocytes and rat hepatocellular carcinoma (hepatoma) cells indicates that hepatoma cells have a higher uptake of palmitate than primary cells but reduced levels of β-oxidation, most likely due to their proliferative nature and requirement for synthesis of new membrane phospholipids (9). In vitro studies of human (HepG2) and rat (H4IIE) hepatoma cells indicate that palmitate treatment leads to numerous lipotoxic events such as reactive oxygen species production (10), insulin resistance (10, 11), endoplasmic reticulum stress (12), and secretion of the pro-inflammatory cytokines interleukin-6 (IL6) and IL8 (13, 14).
Indeed, the combination of cytokines present in an obese or cancer state dictate the broad range of inflammatory responses issued by immune cells and resident tissue cells. Once HCC is initiated, a unique tumor microenvironment develops which is enriched in fibroblasts, blood vessels, stellate cells, extracellular matrix components, and a complex ‘storm’ of cytokines and enzymes secreted from both cancerous and non-cancerous cells. The crosstalk between cytokines differentially recruits myeloid cells such as macrophages and dendritic cells, and influences how the tumor progresses (15). The cytokines released in the tumor microenvironment do not just recruit immune cells from distant sites, however, since evidence suggests that tumor-secreted factors, such as amphiregulin, insulin-like growth factor-II, and IL1, -6, -8, and -18, act as autocrine signals to promote the proliferative and migratory capacity of the tumor cells themselves (16).
As the number of HCC cases is expected to rise with the increasing incidence of metabolic disease, the goal of this study was to determine if the lipotoxicity associated with metabolic syndrome induces an autocrine signaling loop that promotes HCC progression and metastasis.
Materials and Methods
Cell culture and treatment media. H4IIE rat hepatoma cells were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM; ThermoFisher, Frederick, MD, USA), 10% fetal bovine serum (FBS), and penicillin/streptomycin. In the absence of glucose, cells were cultured in glucose-deficient DMEM (ThermoFisher), 2 mM glutamine, 10% FBS, and penicillin/streptomycin. Mouse AML-12 hepatocytes were cultured in DMEM/F12 (ThermoFisher), 10% FBS, 0.1% insulin-transferrin-selenium (ThermoFisher), and penicillin/streptomycin. The fatty acid treatment solutions were prepared by dissolving palmitic acid (Sigma, Saint Louis, MO, USA) or oleic acid (Sigma) in 200-proof ethanol to a final concentration of 195 mM at 37°C. This stock solution was added to 10% fatty acid-free bovine serum albumin (BSA) (Sigma) in phosphate-buffered saline (PBS) at 37°C for a final concentration of 3 mM. Fatty acids were complexed and fully dissolved in 10% BSA for 10 min at 37°C. The final fatty acid:BSA ratio was 2:1. Fatty acid/BSA complexes were diluted in growth media to reach desired treatment concentrations. Ethanol concentrations in media were <0.2%.
Cytokine array and quantitative enzyme-linked immunosorbent assay (ELISA). To screen for a large number of secreted cytokines, cells were plated in 12-well plates. After reaching 75% confluence, growth media was removed from cell monolayers and replaced with 1 ml fatty acid/BSA in growth media. After 24 h, supernatants were removed and centrifuged at 3500 × g for 3 min to pellet floating cells and debris. The entire supernatant was then used in the Proteome Profiler Cytokine Array Kit (Panel A; Bio-Techne, Minneapolis, MN, USA). When performing quantitative ELISAs, cells were plated in 24-well plates. At the time of treatment, growth media were removed and replaced with fatty acid/BSA in growth media, or control media containing BSA mixed with an equivalent volume of ethanol used to solubilize the fatty acids. During harvest, supernatants were removed and centrifuged as above. Supernatants were diluted two-fold (C-X-C motif chemokine ligand 1, CXCL1) or 4-fold (vascular endothelial growth factor, VEGF) and used in the Quantikine ELISA kits (Bio-Techne).
Quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR). H4IIE cells were plated at 2×106 cells per T25 flask. At the time of treatment, growth media was removed and replaced with 400 μM palmitate/BSA in growth media, or control media. Six h after treatment, cells were washed with PBS, and lysed in 0.6 ml lysis buffer with β-mercaptoethanol (ThermoFisher). RNA samples were digested with RQ1 RNase-Free DNase for 15 min at room temperature (Promega) and re-purified. One hundred nanograms of RNA was added to a 20 μl RT-qPCR reaction using GoTaq 1-Step RT-qPCR (Promega) and primers for CXCL1 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) [for primer sequences see (7) and (17)]. Real-time analysis was performed using the SYBR-490 settings on a Bio-Rad iCycleriQ (Hercules, CA, USA). Quantitative measurements were made using the threshold cycle (Ct) value, normalized using the Ct for the reference gene, GAPDH, and analyzed using the 2−ΔΔCt method.
Flow cytometry. After a 6-h treatment in 6-well plates, cells were collected by trypsinization and washed with either PBS plus 0.5% FBS [G-protein-coupled receptor C-X-C motif chemokine receptor 2 (CXCR2)] or annexin V binding buffer (Biolegend, San Diego, CA, USA). Cells were stained with 10 μl mouse IgG2B-allophycocyanin (APC) isotype control (Bio-Techne) or 10 μl mouse anti-rat CXCR2-APC (Bio-Techne) in 100 μl of PBS plus 0.5% FBS, or 5 μl of annexin V-APC or annexin V-fluorescein isothiocyanate (Biolegend) in 100 μl of annexin V binding buffer for 30 min at room temperature. Following staining, cells were washed and resuspended in 2 ml of staining buffer before passing through a 50 μm filter and running on a Partec CyFlow Space MI flow cytometer (Sysmex, Lincolnshire, IL, USA).
Migration assay. H4IIE cells were collected from growth flasks by trypsinization and washed once with PBS plus 0.5% FBS. Cells were labeled in 1 ml PBS plus 0.5% FBS containing 3 μM calcein for 30 min and vortexed every 10 min. Labeled cells were washed once with PBS plus 0.5% FBS and resuspended in the same buffer to 1.5×106 cells/ml. To the upper reservoirs of 8.0 μm pore 24-well Corning® Transwell® chambers (Sigma), 200 μl of the cell solution (3×105 cells) was added; recombinant rat CXCL1 (Bio-Techne) in PBS plus 0.5% or equivalent dilutions of the buffer used to reconstitute lyophilized recombinant protein were added to the lower reservoirs. Assembled migration chambers were incubated at 37°C/5% CO2 for 4 h. At the time of harvest, migration inserts were removed, and fluorescence in the bottom chamber was read (485 nm/520 nm).
Statistical analysis. In this study, data are the result of at least three independent experiments and are shown as the mean±standard error of the mean (SEM), or, when appropriate, the propagation of error using SEM. The significant differences between groups were calculated by a one- or two-sample t-test assuming unequal variances. Differences with a value of p<0.05 were considered as statistically significant.
Results
Palmitate treatment of hepatocytes and hepatoma cells results in different cytokine secretion ‘signatures’. With the goal of surveying a significant number of secreted cytokines from rat hepatoma cells (H4IIE) in response to treatment with 400 μM palmitate, an antibody array was utilized. This concentration of palmitate is relevant since serum FFAs rise above 1 mM after eating a fatty meal or during fasting, and between 30-35% of serum FFAs is palmitate (11). Out of the 29 cytokines available for detection, only two cytokines were detected after a 24-h treatment: VEGF and CXCL1 (Figure 1A). The secretion of these cytokines from cancer cells is consistent with studies that have demonstrated their role in the progression of several cancer types. In patients with colorectal cancer, increased levels of serum VEGF and CXCL1 were found to correlate with metastatic potential (18) and levels of CXCL1 in tumors of patients with HCC correlated with serum alpha-fetoprotein and the risk of tumor recurrence following hepatectomy (19).

Palmitate induces secretion of different cytokines from cancerous compared with non-cancerous hepatocytes. Antibody arrays of supernatants collected from H4IIE hepatoma (A) and AML-12 hepatocyte (B) monolayers after 24 h of treatment with 400 μM palmitate. Spots in the corners of the arrays are positive and negative controls. CCL2: Chemokine (C-C motif) ligand 2; CXCL1/10/12: C-X-C motif ligand-1/-10/-12; IFNγ: interferon gamma; IL1Ra: interleukin-1 receptor antagonist; M-CSF: macrophage colony-stimulating factor; TIMP1: tissue inhibitor of metalloproteinases 1; VEGF: vascular endothelial growth factor.

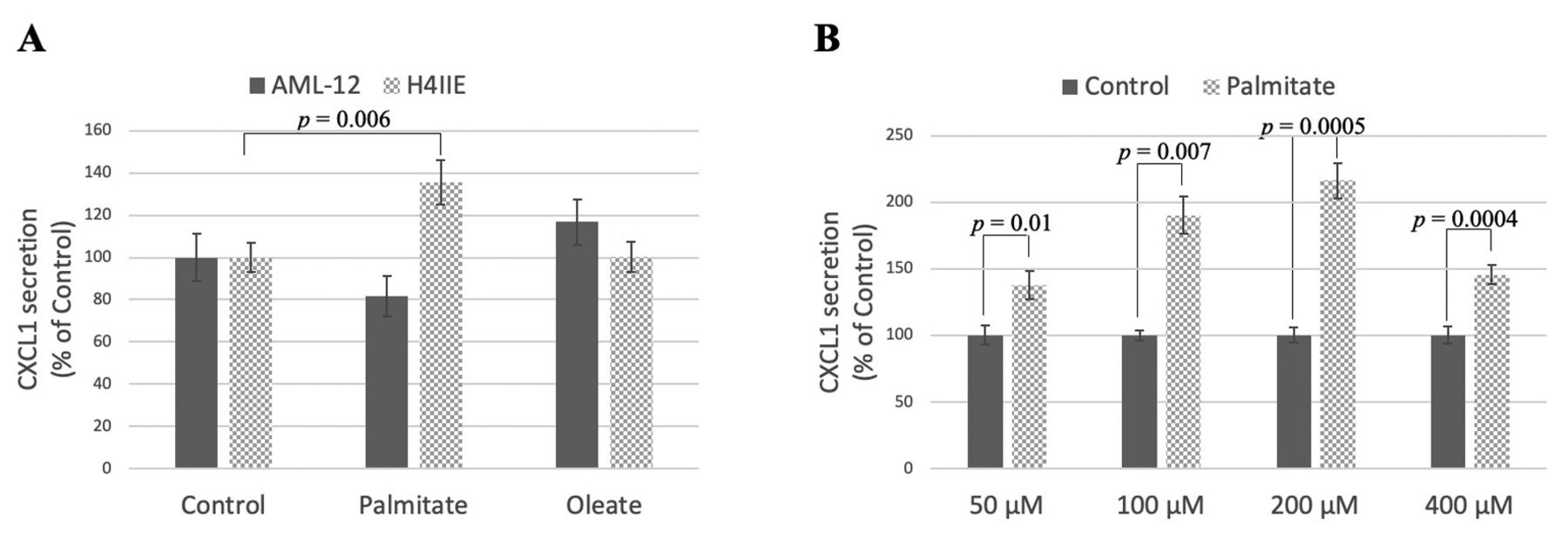
Palmitate induces secretion of C-X-C motif ligand 1 (CXCL1) from hepatoma cells in a dose-dependent manner. A: H4IIE hepatoma cells and AML-12 hepatocytes were treated with 400 μM palmitate/bovine serum albumin (BSA), 400 μM oleate/BSA, or control (ethanol)/BSA for 24 h. The amount of CXCL1 was measured by quantitative enzyme-linked immunosorbent assay (ELISA). B: H4IIE cells were treated with differing concentrations of palmitate/BSA or control/BSA for 24 h. CXCL1 was measured by quantitative ELISA.
In contrast, when the mouse hepatocyte cell line AML-12 was treated with palmitate, a different cytokine profile was observed from a possible 40 cytokines. These non-cancerous cells secreted primarily CXCL1, CXCL10, chemokine (C-C motif) ligand 2 (CCL2), tissue inhibitor of metalloproteinases 1 (TIMP1), and macrophage colony-stimulating factor (M-CSF) with lower levels of interferon gamma (IFNγ), interleukin-1 receptor antagonist (IL1Ra), and CXCL12 also being detected (Figure 1B).
Palmitate dose-dependently induces CXCL1 secretion in hepatoma cells. To confirm the antibody array result, we treated rat hepatoma cells with palmitate and collected cell supernatants after 24 h. When compared to control hepatoma cells, palmitate led to a ~40% increase in CXCL1 secretion (Figure 2A), while no change in VEGF secretion was found (data not shown). This effect was not seen with the unsaturated fatty acid oleate, nor was it observed in non-cancerous AML-12 hepatocytes. The increase in CXCL1 secretion was maximal at 200 μM palmitate (Figure 2B) and was detected as early as 2 h before leveling off at 6 h (data not shown). It is likely that CXCL1 secretion levels plateaued at 6 h because at this time point, 34.6±3.1% (p=0.003 vs. control) of the cells were undergoing apoptosis (Figure 3A and B). This lipotoxicity is likely due to the preference of these cancer cells for glucose as a metabolic fuel, while the uptake of saturated fatty acids in these proliferative cells is used for phospholipid synthesis, resulting in membrane destabilization and endoplasmic reticulum stress (12). We also confirmed the increase in CXCL1 due to palmitate treatment after 6 h at the mRNA level (5.7±0.9-fold change above control, p=0.04).
Palmitate-induced CXCL1 secretion persists in the absence of glucose. By removing glucose from our hepatoma cell culture, we sought to encourage these cancer cells to metabolize the supplied palmitate through β-oxidation, thus reducing the effect of palmitate-induced lipotoxicity. In previous studies, increasing fatty acid β-oxidation reduced pro-inflammatory cytokine secretion in hypothalamic neurons and reduced palmitate-induced apoptosis in C2C12 myotubes (20, 21). As stated previously, in the presence of both palmitate and glucose, 34.6±3.1% of the cells were apoptotic after 6 h; however, in the absence of glucose, the percentage of apoptotic cells decreased to the level of the control, 6.7±1.3%, in the presence of palmitate (Figure 3A and B). Yet the persistence of elevated CXCL1 secretion in these cells suggests that CXCL1 release is not due to a non-specific lipotoxic response but that palmitate or a direct metabolite is responsible (Figure 3C).
Hepatoma cells express CXCR2 and migrate in response to CXCL1. CXCL1 production is known to both recruit neutrophils to sites of inflammation and induce pathogenic activity by binding to CXCR2 (22). In situ investigation of CXCR2 expression in tumor tissue from patients with HCC determined that the majority of CXCR2+ cells were indeed CD15+ neutrophils, followed by CD3+ T-cells, CD68+ macrophages, and S100+ dendritic cells (23). Besides immune cell CXCR2 expression, Liu et al. reported an increase in CXCR2 expression level in human HCC tumor cells compared to surrounding liver cells (24). To determine if secreted CXCL1 can stimulate an autocrine signaling axis, the goal of our study was to measure the level of CXCR2 expression on the surface of hepatoma cells both at baseline and under palmitate-treated conditions. By flow cytometry, we detected a 4.5-fold increase in CXCR2 expression following 6-h treatment with 400 μM palmitate (Figure 4A). This increase in expression occurred primarily within the highly apoptotic cell population as observed when co-stained with annexin V (Figure 4B). We hypothesize that CXCL1 binds to CXCR2 on the surface of apoptotic cells to promote signaling events that lead to rescue of these cells from apoptosis or migration from the lipotoxic environment. As such, CXCL1 acting through CXCR2 has been shown to protect astrocytes from ischemia-induced apoptosis by preventing mitochondrial cytochrome c release (25). A recent study demonstrated that the ability of cells to recover from apoptosis, termed anastasis, may be detrimental when occurring in cancer cells (26). The escape of a tumor from chemotherapy-induced apoptosis may lead cells to acquire new and beneficial DNA mutations during survival and subsequent proliferation.
In addition to the possibility of CXCL1-stimulated anastasis, we wanted to determine if the expression of CXCR2 on H4IIE cells increases their own migratory capacity. When 100 pg/ml of recombinant rat CXCL1 was placed in the lower chamber of a Transwell assay, we observed a ~60% increase in the number of migrating hepatoma cells from the top chamber, with the number of migrating cells increasing further when 250 pg/ml CXCL1 was placed in the lower chamber (Figure 4C). Thus, signaling through CXCR2 binding likely causes cytoskeletal changes that promote migration in hepatoma cells. These results suggest that even a small increase in CXCL1 secretion due to palmitate, like that shown in Figure 2, can have a significant effect within the local tumor environment. Secreted CXCL1 from cells exposed to palmitate may stimulate self or neighboring hepatoma cells to recover from apoptosis or influence metastatic potential by promoting migration.
Discussion
As obesity is associated with up to 20% of all cancer-related deaths in the U.S. (27), nutritional interventions are currently being explored for cancer prevention, treatment, and tolerance of chemotherapeutics. Unlike lung, breast, and colonic cancer, which arise in a healthy tissue environment, HCC develops in the context of underlying tissue steatosis. Therefore, HCC is particularly heterogeneous as the genome instability regularly seen in cancer is compounded by the epigenetic modifications that result from an individual's dietary composition (28). This heterogeneity complicates the comparison of studies in patients with cancer and in vivo models and validates the need for information obtained from in vitro cell culture models.
Caution must be used, however, when interpreting data from different laboratories as the clear reporting of FA delivery methods is imperative when comparing in vitro results. Cell culture experiments demonstrate that palmitate is more efficiently taken up by cells in the presence of the amphipathic protein albumin (29). Ensuring the albumin source is not contaminated or denatured by high temperatures during FA complexing procedures is crucial for studies investigating inflammation. In healthy humans, the ratio of FA binding to serum albumin is between 1:1 and 3:1 but this ratio can increase in disease states. While the use of BSA is a good substitute for human albumin, differences exist in particular FA binding affinities (30). We chose a 2:1 ratio to minimize levels of unbound FA and, instead, focus on the direct activation of signaling processes by the specific FA.
In individuals with metabolic syndrome or obesity, developing HCC is exposed to palmitate from multiple sources: Dietary FAs in chylomicron remnants, lipolysis occurring in surplus adipose tissue, and increased de novo lipogenesis as a result of excess dietary carbohydrates. As reviewed elsewhere (31), palmitate can be taken up by cells through simple diffusion or facilitated diffusion using transporters such as CD36 or fatty acid transport proteins. Once in the cytosol, it interacts with fatty acid binding proteins and is chaperoned around the cell, where it can be utilized as a substrate for numerous enzymes or act as a transcription factor binding partner. Alternatively, palmitate may induce intracellular signaling cascades by binding to cell-surface receptors such as toll-like receptor 4 or G-protein coupled receptor 120 (also known as FFAR4) which influence the activity of nuclear factor kappa B (32, 33).
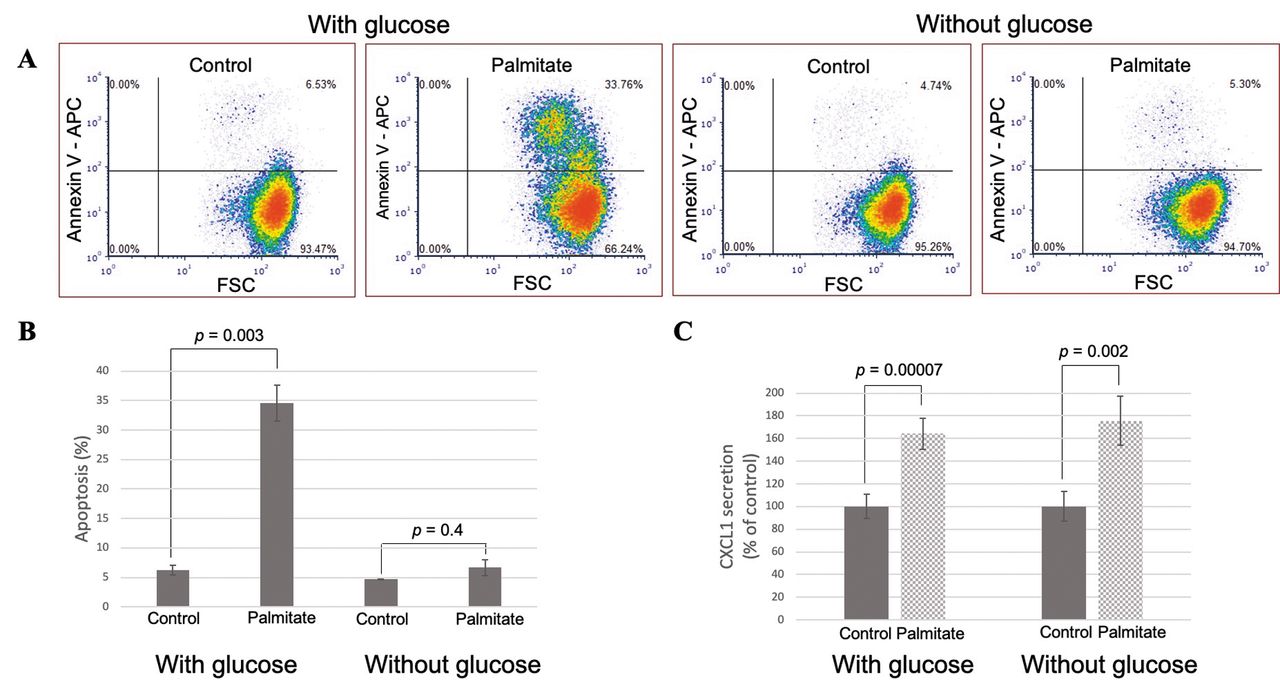
Palmitate induces secretion of C-X-C motif ligand 1 (CXCL1) under conditions when lipotoxicity is reduced. A: H4IIE cells were treated with 400 μM palmitate/bovine serum albumin (BSA) or control/BSA in the presence or absence of glucose for 6 h, then stained with annexin-V for flow cytometric analysis. Dot plots are shown from a representative experiment, with summary and statistical data illustrated in (B). C: H4IIE cells were treated with 400 μM palmitate/BSA or control/BSA in the presence or absence of glucose for 6 h and supernatants were collected and assayed for CXCL1 secretion by quantitative enzyme-linked immunosorbent assay (ELISA).
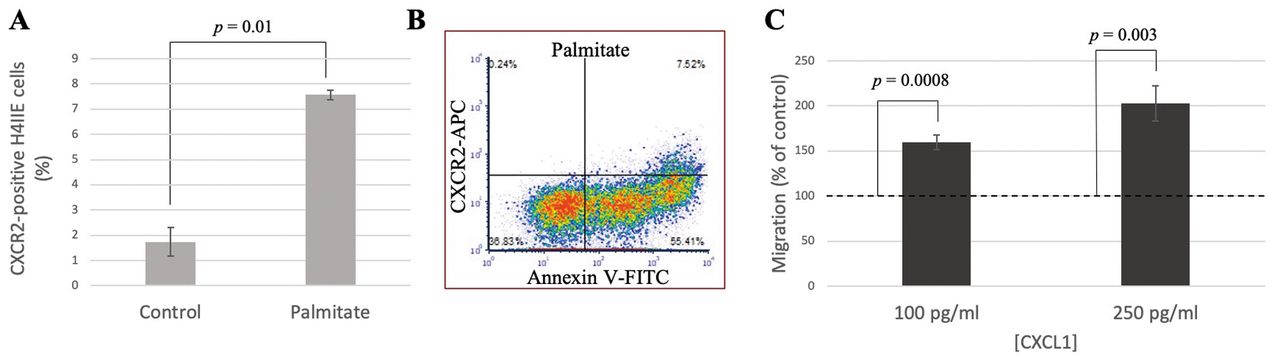
C-X-C motif chemokine receptor 2 (CXCR2) expression is increased on apoptotic cells and promotes migration of hepatoma cells. H4IIE cells were treated with 400 μM palmitate/bovine serum albumin (BSA) or control/BSA and stained with anti-rat CXCR2-allophycocyanin (APC) with (B) and without (A) annexin V-fluorescein isothiocyanate (FITC). A dot plot is shown from a representative experiment. C: In vitro migration assay of H4IIE cells towards recombinant rat C-X-C motif ligand 1 (CXCL1).
Other studies of lipid-induced cytokine secretion fail to mention apoptosis that is likely occurring in the cells, although its observation should not be ignored. Increased apoptosis in the liver results in the release of apoptotic bodies which can be engulfed by both hepatic stellate cells and Kupffer cells, promoting further inflammation and fibrosis (34). Furthermore, increased apoptosis paradoxically correlates with tumor recurrence as growth signals are released from apoptotic tumor cells, leading to additional oncogenic mutation and proliferation of neighboring live tumor cells (35).
As CXCL1 is the only cytokine whose levels changed in response to palmitate, it is a good candidate for cancer treatment. Therapy with respect to CXCL1 and CXCR2 has been explored, however, not in the context of nutritional or metabolic modification. In xenograft models, reduced tumor size was observed either when CXCL1 was knocked down by small interfering RNA in HCC cells (36), or when animals were treated with therapeutic T-cells transduced with CXCR2 (37). In conclusion, we demonstrate here a role for palmitate in the up-regulation of an autocrine signaling axis involving CXCL1 and CXCR2. While both autocrine and paracrine networks are likely at play in the full context of cancer, the management of HCC lipotoxicity due to metabolic syndrome and obesity is important for the prevention of metastasis and chemotherapy resistance.
Acknowledgements
The Authors would like to thank the 2019 and 2020 Biochemistry II classes at Bethel University; Abby Puk, Clayton Neuenschwander, and Brooke Toutloff for antibody array results; Jamey Young for the generous gift of H4IIE cells; Yonglong Zou and Aaron Boeckermann for assistance with the migration assay; Andrew Zieffler and Wade Neiwert for help with data analysis; and Laura Listenberger for careful reading of the article. This work was supported in part by a Sigma Zeta National Science & Mathematics Honor Society Student Research Award.
Footnotes
Authors' Contributions
AKS was responsible for study design, project supervision, and writing the article. KJVD, LCV, and AJF were responsible for conducting experiments and data analysis.
This article is freely accessible online.
Conflicts of Interest
The Authors declare no conflicts of interest.
- Received August 22, 2020.
- Revision received September 4, 2020.
- Accepted September 8, 2020.
- Copyright© 2020, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}