


Abstract
Background/Aim: Malignant melanoma is the most aggressive type of skin cancer, with increasing frequency and mortality. Melanoma is characterized by rapid proliferation and metastases. Malignant transformation of normal melanocytes is associated with imbalance between oncogenes' action and tumor suppressor genes. Mutations or inactivation of these genes plays an important role in the pathogenesis of malignant melanoma. Many target-specific agents improved progression-free survival but unfortunately metastatic melanoma remains incurable, so new therapeutic strategies are needed. The balance of histones' acetylation affects cell cycle progression, differentiation and apoptosis. Histone deacetylases (HDAC) are associated with different types of cancer. Histone deacetylase inhibitors (HDACI) are enzymes that inhibit the action of HDAC, resulting in block of tumor cell proliferation. A small number of these enzymes has been studied regarding their anticancer effects in melanoma. The purpose of this article was to review the therapeutic effect of HDACI against malignant melanoma, enlightening the molecular mechanisms of their action. Materials and Methods: The MEDLINE database was used. The keywords/ phrases were; HDACI, melanoma, targeted therapies for melanoma. Our final conclusions were based on studies that didn't refer solely to melanoma due to their wider experimental data. Thirty-two articles were selected from the total number of the search's results. Only English articles published until March 2017 were used. Results: Molecules, such as valproid acid (VPA), LBH589, LAQ824 (dacinostat), vorinostat, tubacin, sirtinol and tx-527, suberoyl bis-hydroxamic acid (SBHA), depsipeptide and Trichostatin A (TSA) have shown promising antineoplastic effects against melanoma. Conclusion: HDACI represent a promising agent for targeted therapy. More trials are required.
Malignant melanoma is the most aggressive type of skin cancer. It arises from melanocytes and spreads via the bloodstream and the lymphatic system. It emerges in the skin and the mucous membranes of the mouth and genitals (1). Among melanoma cases, 90% afford the skin and 10% the eyeball, the brain meninges, the mucous membrane of the oral cavity and the genitalia (2). The frequency of melanoma is constantly increasing worldwide and mortality is rising faster than any other form of cancer (3).
Melanoma is characterized by rapid proliferation and numerous metastases. Malignant transformation of the normal melanocytes is associated with the imbalance between the action of oncogenes and tumor suppressor genes. Mutations or inactivation of these genes plays an important role in the pathogenesis of the malignant melanoma (1).
In recent years many target-specific agents [i.e. BRAF inhibitors, Histone deacetylase inhibitors (HDACI)] have been developed and have improved progression-free survival but unfortunately metastatic melanoma is still incurable. A variety of factors have been associated with resistance to therapy such as tumor cell alterations during tumor progression and the microenvironment including chemokines (4).
Recent studies have shown that BRAF mutations affect melanoma cell survival through the MAPK pathway. Especially MAPK activation leads to transcriptional changes of the DNA which have an important role in melanoma cell differentiation and proliferation. Almost 40% of cutaneous melanoma patients have tumors with BRAF mutations which activate the MAPK pathway through phosphorylation that activates MEK and ERK genes. Phosphorylation of these genes leads to enhanced cell proliferation and survival signals (5).
Histone deacetylase (HDAC) proteins get involved in the malignant transformation by affecting the chromatin structure and the transcription of several genes that are associated with the cell cycle. HDACI are pharmacologic compounds that interfere with the deacetylation process mediated by HDAC, leading to a global increase of histone acetylation. HDACI represent promising therapeutic agents because they can affect the survival of tumor cells and their environment (4).
HDAC Classification
HDAC comprise of 18 different substances subdivided into four classes: class I consists of HDAC-1, -2, -3 and -8 and are located in the cell nucleus. Class II consists of HDAC-4, -5, -6, -7, -9 and -10 that are characterized by exonuclear and nuclear localization. Class III includes sirtuins (SRT-1-7 genes) and Class IV consists of only HDAC-11 which has characteristics of both Class I and II. Classes I, II and IV of HDAC require a zinc (Zn+) ion to transfer acetyl groups from their substrates. Sirtuins have no similarities with the other three groups of HDAC and require a nicotinamide adenine dinucleotide (NAD+) ion for their catalytic activity (6) (Table I).
Classification of histone deacetylases.
HDAC Mechanism of Action
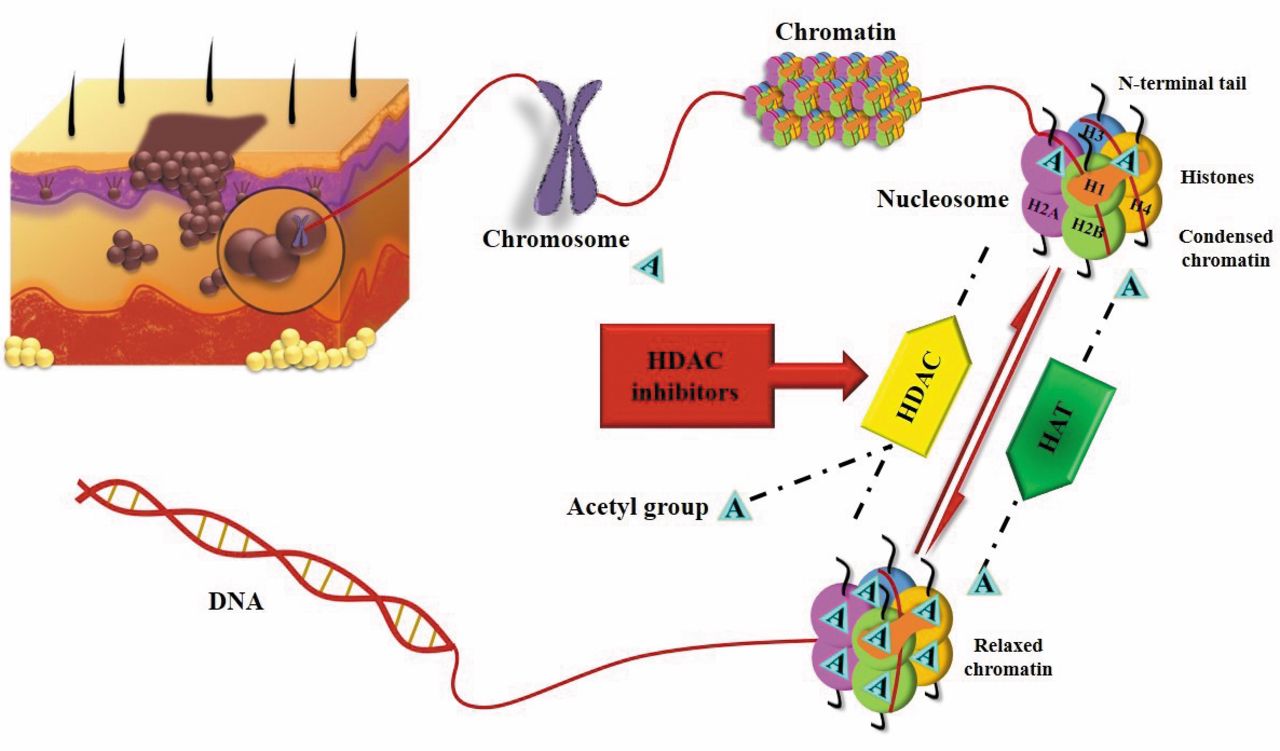
The role of HDAC is opposite to the activity of histone acetyltransferases (HAT). The balance between the action of HDAC and HAT define the level of acetylation of the histones. HDAC catalyze the removal of the acetyl group on lysine residues of proteins and nucleosome histones H2A, H2B, H3 and H4 are some of the substrates of HDAC. The balance of acetylation of histones, regulates the chromatin structure and as a consequence the transcription of many genes. Acetylation of lysine residues neutralizes the histones and leads to a less condensed chromatin structure by increasing the space between nucleosome and the DNA wrapped around it (Figure 1). As a result of this change to the chromatin structure, transcription factors can get combined with the DNA in such a way that it can activate the transcription of many genes that get involved in the control of the cell cycle progression, differentiation and apoptosis (5, 6). Moreover, it has been shown that other acetylated proteins are substrates of HDAC, including NF-YA, p53 and GATA-1 (6). HDAC have been associated with different types of cancer. Some of them include over-expression of HDAC-1 and -5 associated with gastric cancer, HDAC-2 and -3 have been found in colorectal cancer and SIRT-8 is over-expressed in thyroid cancer (6, 7). In addition, HDAC-1, -2 and -3 have been associated with high levels of activated NF-kB which result in poor prognosis in patients with pancreatic cancer (7).
Anti-cancer Action of HDACI in Melanoma
HDACI are enzymes that inhibit the action of HDAC causing hyperacetylation of their substrates (Figure 1). Extensive research has shown that HDACI can block tumor cell proliferation and differentiation by affecting the balance of histone acetylation. There are four main classes of HDACI that differ regarding their structure, hydroxamic acid, cyclic tetrapeptide, benzamide and aliphatic acid (6). Although there is a big variety of these enzymes, a small number has been studied about their anticancer action in melanoma.
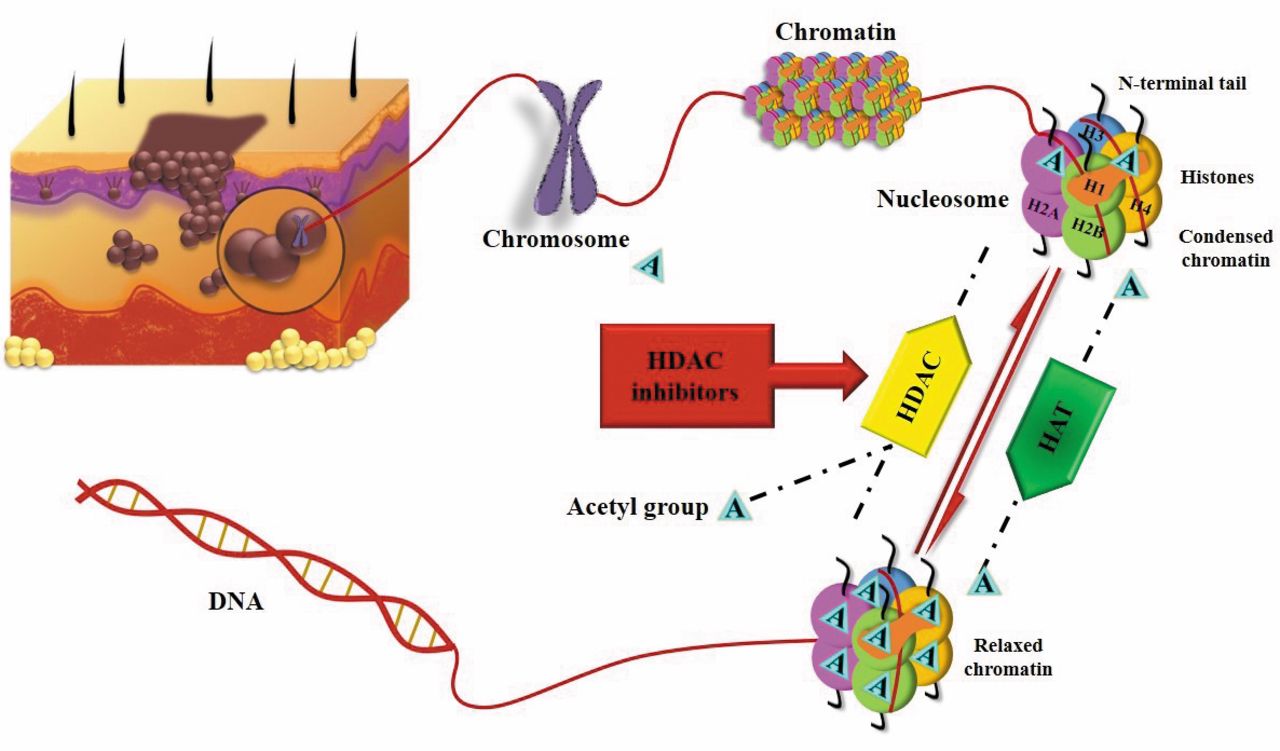
Mechanism of action of histone deacetylase inhibitors (HDACI) as targeted therapy against malignant melanoma. Histone acetyl transferases (HAT) catalyze lysine acetylation; Histone deacetylases (HDAC) remove acetyl groups. HAT: Histone acetyltransferases; HDAC: histone deacetylases.
Extensive research has shown that acetylation of many genes takes place during melanogenesis and this reaction contributes to the down-regulation of the tumor suppressor genes such as p14ARF and p16INK4a (6). Apart from the histones, HSP90, p53, NF-kB and the subunit of p65 are also targets of HDAC proteins.
Studies on the HDAC function, have shown that hyperacetylation of protein HSP90 following HDAC inhibition causes: a) the degradation of C-RAF and Akt signaling molecules which have an important role as mediators of growth and b) degradation of the upstream receptor tyrosine kinases ERBB1 and ERBB2 that is associated with microtubule affinity regulating kinase (MARK) inhibitor resistance.
In addition, alteration of p53 activity and activation of NF-kB may alter melanoma resistance to therapy and may also lead in differentiation in cytokine production, transcription of anti-apoptotic protein and immune response (1, 6). Table II summarizes the major actions of HDACI against malignant melanoma.
VPA. Induction of apoptosis is a significant effect of valproic acid (VPA), an extensively studied HDACI. VPA increases the activity of two of the caspase-3, by inhibiting the activity of two of the four classes of HDAC, I and II, except for HDAC-9 and -11 which are activated by VPA (1). Caspase-3 belongs to cysteine proteases ICE and participates in the apoptosis procedure. VPA also inhibits angiogenesis and metastasis inducing the cell cycle arrest in G1 phase. This action of valproic acid is achieved with up-regulation of p16 protein, a cell-cycle inhibitor (2). It has also been shown that pan-HDACI enhancing apoptosis in malignancy with up-regulation of BIM, BAX and BIK, proteins with apoptotic action and with down-regulation of Bcl-XL and XIAP which have anti-apoptotic action (8).
HDAC-9 and -11 are activated by VPA (1). Caspase-3 is involved in the apoptosis and it can be activated by depsipeptide and SAHA (9, 10). Besides VPA inhibits angiogenesis and metastasis inducing the cell cycle arrest in G1 phase. This action of valproic acid is achieved with up-regulation of the p16 protein, a cell-cycle inhibitor (2). It has also been shown that pan-HDACI enhance apoptosis in malignancy with up-regulation of BIM, BAX and BIK, proteins with apoptotic action and with down-regulation of Bcl-XL and XIAP which have anti-apoptotic action (8, 10-12).
LBH589. It has been found that low nanomolar concentrations of LBH589 inhibit the growth of all melanoma cell lines tested, but not normal melanocytes. This inhibition is characterized by increased apoptosis as well as a G1 cell cycle arrest. In addition, LBH589 augments the expression of MHC and co-stimulatory molecules on melanoma cells leading to an increased ability to activate antigen specific T-cells. In vivo, LBH589 treatment of melanoma-bearing mice results in a significant increase in survival (13).
LAQ824. The HDACI LAQ824 has antitumor activity in melanoma tumors, but the degree of induced apoptosis may vary. It induces the expression of p21, increases production of reactive oxygen species and activates the caspase cascade (14). When combined with retinoids, therapeutic results are promising.
Major actions of histone deacetylase inhibitors against malignant melanoma.
Vorinostat. The safety and efficacy of the hydroxamate vorinostat (SAHA) were assessed in combination with the proteasome inhibitor marizomib in melanoma, pancreatic cancer and lung cancer in a phase I study. The efficacy of both agents was assessed in vitro in tumor cell lines revealing a marked synergy in terms of growth inhibition and induction of apoptosis (15).
Tubacin. HDAC-6 inhibition with tubacin results in the accumulation of acetylated α-tubulin, but not acetylated histones in normal human foreskin fibroblast (HFS) cells and transformed human prostate cancer (LNCaP) cells. Tubacin reduces the rate of growth of transformed and, to a lesser extent, normal cells, without loss of cell viability (16).
Sirtinol and tx-527. SIRT-1 is a member of the class III HDAC known as the Sirtuins, which depend on nicotinamide adenine dinucleotide (NADC) as a substrate. Wilking et al. (17), studied the action of SIRT-1 inhibitors and found that the inhibition of SRT1 decreases the phosphorylation of PIP2 in PIP3 by inhibiting PI3K so the cell migration is limited (13). SIRT-1 inhibitors can increase the levels of p53 and p21, two proteins which are important for the cell-cycle arrest to inhibit the proliferation of tumor cells (17). The role of SIRT-1 in melanoma is beginning to be unraveled only recently and SIRT-1 inhibition seem to affect p53 protein levels in wild-type p53 melanoma cells, while several other pathways may be affected by SIRT-1 inhibition, including a possible role in cell-cycle regulation. Other studies have shown that the combination of pan-HDACI SAHA and termozolamide might have a therapeutic advantage in melanoma treatment by causing apoptosis in tumor cells. The combination of these two drugs especially, is responsible for down-regulation of chemokine (C-C motif) ligand 2 (CCL 2), which is associated with melanoma growth and proliferation (4).
SBHA. The new HDACI suberic bishydroxamate (SBHA), is responsible for its effect on a panel of human melanoma cell lines. It induces varying degrees of apoptosis in the melanoma lines but not in melanocytes and fibroblasts. Induction of apoptosis is caspase dependent and associated with induction of changes in mitochondrial membrane permeability, which could be inhibited by overexpression of Bcl-2. SBHA down-regulates several key antiapoptotic proteins including X-linked inhibitor of apoptosis and the Bcl-2 family proteins, Bcl-XL and Mcl-1. In contrast, it induces up regulation of the Bcl-2 family proapoptotic proteins, Bim, Bax, and Bak (18). In the study of Gillespie et al., TRAIL and SBHA synergistically induce apoptosis of melanoma cells as revealed by quantitative analysis using the normalized isobologram method. This is supported by enhanced activation of caspase-3 and cleavage of its substrates, PARP and ICAD (19).
Despipeptide. Another study approved that in the presence of despipeptide, a well-known HDACI, B16/F10 melanoma cells induced apoptotic cell death after Fas-FasL cross-linking, something that does not happen with Fas cross-linking alone (9, 20). Besides, depsipeptide can increase the perforin expressing CTLs and perforin in conjunction with granzyme induces an apoptotic cascade in melanoma cells (9). Another study has proved that HDACI LAQ 824 in combination with retinoic acid can increase the production of ROS, leading to the activation of caspase cascade and degradation of Bcl-2 anti-apoptotic protein in melanoma cells (14). Antineoplastic action of HDACI is related with the prevention of transcription (9, 21). Greer et al. showed that HDACI can promote the connection between RNAP2 and negative elongation factor (NELF), which blocks elongation by preventing the action of RNAP2. The same research revealed that HDACI can prevent the transcription of many genes with an additional mechanism: HDACI gets associated with the redistribution of bromodomain-containing protein 4 (BRD-4) which recruits the positive-transcription elongation factor b (P-T EFb). P-T EFb has an important role in the transcription of many genes because it modifies RNAP2 and other factors required for overcoming elongation block (21).
Trichostatin A (TSA). Trichostatin is a “non-specific” HDACI. It increases expression of the cyclin-dependent kinase inhibitor (CDKI) p21, while it down-regulates p53. It causes reduction of cyclin D1 and cyclin A protein (10) and reduces the levels of Bcl-2 and Bcl-XL anti-apoptotic proteins. It also increases the expression of Il-2, TNF-a, GM-CSF and the activation of T-cells. Moreover, TSA induces autophagy in cells through inhibition of rapamycin motor pathway (22) and inhibits apoptosis of activated CD4+ T lymphocytes (23). Butyrate as well as trichostatin A inhibited the cell growth mainly by arresting the cell cycle and the cell invasion was inhibited by butyrate and trichostatin A. At low doses, sodium butyrate and trichostatin induced a G1 cell-cycle block in melanoma cells (23).
Discussion
It is speculated that HDACI sensitize melanoma cells to immunotherapy. Malignant melanomas in situ contain a high level of HDAC-1, 2 and malignant melanoma cells overexpress HDAC-1, -2, -3 compared with non-cancer cells. Furthermore, pharmacologic inhibition of class I HDAC sensitizes malignant melanoma cells to apoptosis following exposure to alkylating agents, while not affecting primary melanocytes (24-26).
West et al. (27) showed that the immune system is absolutely necessary for the anticancer effect of HDACI (9, 11, 24, 28). HDACI distribute to the up-regulation of natural killer NK1-cell activating ligands MHC class I and II, which are responsible for antigen presentation on the surface of cancer cells (22). HDACI can sensitize malignant cells to the antineoplastic effect of IFN-γ by increasing the number of IFN-γ receptors (IFNGR1) on the surface of cancer cells. The same group approved that B cells have a critical role in the anticancer effect of HDACI by producing IFN-γ (27).
Recent micro-array studies revealed that depsipeptide a well-known HDACI, increases gp100 expression on the surface of melanoma cells. This HDAC class I molecule is released from the endoplasmic reticulum reaches the cell surface and this up-regulation enhances the immunological recognition. The same study showed that depsipeptide can increase the action of T-cells in such a way that promotes tumor-specific T-cell mediated killing of melanoma cells (9).
Except for gp100, TAP-1 and TAP-2 have an important role in the recognition of melanoma cells by CD8 CTLs, as they transport peptides processed by the proteasome into the endoplasmic reticulum, where they are loaded onto MHC class II (11). Setiadi et al. showed that HDACI are able to increase the expression of TAP-1 by enhancing recruitment of RNA polymerase II to the TAP promoter so as to promote immune recognition of melanoma (11). Cao et al. showed that HDACI have the possibility to increase the action of the immune system by preventing the activation-induced cell death. In this study TSA was approved, the first FDA-approved HDACI that causes down-regulation of FasL expression on infiltrating CD4+ T cells, to inhibit the apoptosis of CD4+ T lymphocytes. Additionally, the same study showed that TSA and CTLA4 antibody act synergistically to enhance CD4+ T-cell infiltration and that TSA can increase the expression of Il-2, TNF-a and GH-CSF, cytokines associated with cell activation (23).
It is known that HDACI have a great anti-proliferative effect on melanoma cells (6, 9, 10, 12). Borrielo et al. demonstrated that HDACI of class I and II can cause the accumulation of melanoma cells in G1 phase due to the elongation of p27half-life. The p27 is a CDKI, which can mediate G1 arrest in response to an array of stimuli including DNA damage, mitogen deprivation or drug treatment. The level of this protein can be decreased by ubiquitination followed by proteasomal degradation. HDACI have the capability to down-regulate Skp2 protein, a component of the nuclear p27 ubiquitination complex due to translational and post translational mechanism to increase p27half-life (12).
Most HDACI can induce the expression of cyclin-dependent kinase inhibitors (CK1) p21 by increasing acetylation of the chromatin at the sp1 binding site in the promoter region of the gene. High levels of p21 are associated with G1/S phase cell cycle arrest due to the inhibition of CDK2 (10, 29).
Additionally, HDACI can cause down-regulation of TbX2, a protein that represses p21 expression in vitro and in vivo and plays an important role in melanoma proliferation (10, 29). Vance et al. showed that TbX2 is over-expressed in melanoma cells and that HDAC proteins are potential mediators of TbX2 proteins, a fact that can explain the capability of HDACI to cause down-regulation of TbX2 (10, 29, 30).
HDACI can also induce autophagy in melanoma cells through FOXO1-dependent pathways. Forkhead box proteins (FOXOs) are transcription factors which have an important role in regulation of genes involved in cell growth, proliferation and differentiation, with FOXO1 being the most studied (Autophagy is a mechanism which provides cells with additional nutrient supplies under stress conditions and is mediated by the suppression of rapamycin, MTOR, a negative regulator of autophagy) (22, 31). Zhang et al. approved that HDACI can enhance FOXO1 acetylation and transcriptional activation by suppressing the action of deacetylases. This way the transcription of ATGs genes increases (autophagy related genes), which are necessary for the autophagy of cancer cells (22).
Nowadays, several HDACI such as vorinostat, entinostat and valproic acid have recently been tested in phase I and early phase II trials, yet most agents show limited efficacy and tolerability as single agents. Haematological toxicity, fatigue, nausea and laboratory abnormalities are the most frequent adverse events of HDAC inhibition (24, 32).
Conclusion
In recent years many studies have focused on examining the efficacy of HDACI in the treatment of melanoma, with TSA being the most well studied. It has been approved that HDACI play an important role in the restriction of the growth and proliferation of melanoma cells especially by inducing cell cycle arrest and apoptosis with different mechanisms. In addition, the synergistic action of HDACI with other inhibitors such as BRAF inhibitors and BET inhibitors has been studied and has been approved as a quite effective treatment of melanoma but further research is required on this subject. The fact that four HDACI, vorinostat (5AIIA, Zalmza), romidepsine (Istodox, depsipeptide), belinostat and panobinostat (Farydak) have been accepted by the FDA for treatment of CTCL (cutaneous T-cell lymphoma) and PTCL (peripheral T-cell lymphoma) and multiple myeloma means that HDACI are molecules which will be used further in the future for cancer therapy and more research may result in the acceptance of these molecules by the FDA for melanoma treatment.
Footnotes
↵* These Authors contributed equally to this study.
This article is freely accessible online.
- Received July 23, 2017.
- Revision received August 2, 2017.
- Accepted August 4, 2017.
- Copyright© 2017, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue





{kind=link}
Jump to section
Related Articles
Cited By...
- Valproic Acid Enhances Radiosensitization via DNA Double-strand Breaks for Boronophenylalanine-mediated Neutron Capture Therapy in Melanoma Cells
- Histone Deacetylase Inhibitors and Anaplastic Thyroid Carcinoma
- Whole-transcriptomic Profile of SK-MEL-3 Melanoma Cells Treated with the Histone Deacetylase Inhibitor: Trichostatin A
- The Role of Histone Deacetylase Inhibitors in Uveal Melanoma: Current Evidence
- Abdominal Emergencies in Patients with Stage IV Melanoma: The Role of Surgery: A Single-centre Experience