


Abstract
Desmoplastic small round cell tumour (DSRCT) is a rare tumour, usually arising in the abdominal cavity. DSRCT remains an aggressive malignancy, with a poor prognosis despite multi-modality treatments. In the published literature, there has been no patient who lived for three years or more without surgical excision. This report describes a case of DSRCT arising from the brachial plexus and successfully treated with caffeine-assisted chemotherapy. A 29-year-old male presented with pain and numbness in his left forearm. Radiological findings were suggestive of malignant tumour. Histology, immunohistochemical stain and fluorescence in situ hybridisation (FISH) results confirmed the diagnosis of DSRCT. He underwent caffeine-potentiated chemotherapy and the tumour disappeared. The tumour was not removed surgically as it was intertwined in the brachial plexus. Four years after the initial diagnosis, no local relapse and no distant metastases have been observed. Therefore, it is concluded that caffeine-assisted chemotherapy should be one of the treatment options for DSRCT.
Desmoplastic small round cell tumour (DSRCT) is a rare, high-grade malignant tumour. DSRCT is a mesenchymal neoplasm that grows along serosal surfaces, often involving the abdominal and/or pelvic peritoneum of young male patients. DSRCT was first described in 1989 by Gerald and Rosai (1). To date, approximately 200 cases have been described in the English language literature (1-14). Most have occurred in the peritoneal cavity with widespread peritoneal involvement, but other primary sites have been reported, including the paratesticular region, pleural serosa, posterior cranial fossa, soft tissues, bone and ovary. The prognosis remains poor (overall survival at 5 years: 0-15%) and DSRCT leads to death in most cases (15), despite surgical resection, radiotherapy and high-dose chemotherapy. This study presents the first report of DSRCT occurring in the brachial plexus and being successfully treated with caffeine-assisted chemotherapy.
Case Report
A 29-year-old man had experienced a six-year-long history of pain and numbness in his left forearm. The pain and numbness gradually worsened and the biceps muscle began to atrophy. The patient complained of numbness on the radial side of his left forearm and muscle atrophy in his biceps and triceps. Deep tendon reflexes were absent in the left upper limb. Tinel's sign was present at the left supraclavicular fossa. No lesion was shown on plain radiograph or computed tomography exams. Magnetic resonance (MR) imaging revealed a lesion in the left brachial plexus (Figure 1). The mass demonstrated low intensity on T1-weighted images and high intensity on T2-weighted images. Gadolinium-enhanced T1-weighted images showed heterogeneous enhancement. 201Thallium (201Tl) scintigraphy and 99mTc hexakis-2-methoxyisobutyl-isonitrile (99mTc-MIBI) scintigraphy showed accumulation in the left supraclavicular area (Figure 2a, b). Positron-emission tomography (PET) showed high accumulation on the left brachial plexus, but no metastases were observed (Figure 2c). The leading radiological diagnosis was schwannoma, with other possibilities including malignant peripheral nerve sheath tumour and lymphoma.
An open biopsy was performed, and the pathological diagnosis was DSRCT. The specimen was composed of the proliferation of the small round cell having been separated by the fibrotic and desmoplastic stroma (Figure 3a). Immunohistochemistry demonstrated striking positivity for keratin AE1/AE3 (Figure 3b), epithelial membrane antigen (EMA) and desmin in the small cell component (Figure 3c), while that for myf-4 (myogenin) was negative. Stains for CD99, S100, and neuron-specific enolase (NSE) were also negative, but there was nuclear positivity for polyclonal WT-1. A subset of the cells was positive for p53 and MIB-1 (Figure 3d). A disrupted EWS gene was demonstrated by fluorescence in situ hybridisation (FISH) assay (Figure 3e).
After five courses of chemotherapy were performed using cisplatin (120 mg/m2 for 2 h), doxorubicin (30 mg/m2/day for 2 days) and caffeine (1.5 g/m2/day for 3 days), the size of the tumour markedly decreased on MRI (Figure 4), and the accumulation of 201Tl and 99mTc-MIBI disappeared (Figure 5). The tumour was not removed surgically as it was intertwined in the brachial plexus. Three courses of additional chemotherapy were performed using ifosfamide (3 g/m2/day for 3 days), etoposide (60 mg/m2/day for 3 days) and caffeine (1.5 g/m2/day for 3 days), and radiation therapy with 60 Gy in total.
At present, forty-six months after the initial diagnosis, no local relapse and no distant metastases have been observed.
Discussion
DSRCT is a rare, high-grade malignant tumour that usually occurs in males during adolescence and early adulthood. It frequently presents as a large abdominal mass with widespread peritoneal involvement at diagnosis. It has been reported that DSRCT can occasionally involve other body sites such as the lung, salivary gland, ethmoid sinus, kidney, pancreas, soft tissue and the posterior cranial fossa (7, 11, 16, 17). This is the first reported case of a DSRCT arising from brachial plexus. The location helped to detect the tumour before it became enlarged. However, the location made it more difficult to decide on the treatment plan because it was impossible to excise the tumour without sacrificing the nerve.
DSRCT belongs to the family of ‘small round blue cell tumours’, which includes primitive neuroectodermal tumour (PNET), Wilm's tumour and Ewing's sarcoma. Cytologically, it is difficult to differentiate from other small round cell tumours such as primitive neuroectodermal tumour/Ewing's sarcoma, small cell carcinoma, neuroblastoma, Wilms' tumour, lymphoma and rhabdomyosarcoma. Immunohistochemical studies are helpful for the diagnosis of DSRCT. Unlike other small round cell tumours, DSRCT expresses both epithelial and mesenchymal markers on immunohistochemical staining. DSRCT has a characteristic reciprocal chromosome translocation t(11;22)(p13;q12) (18-21), which results in the fusion of the N-terminal region of Ewing's sarcoma gene (EWS), located at 22q12, and the C-terminus of Wilm's tumour gene (WT1), located at 11p13, to produce a tumour-specific fusion protein, EWS-WT1 (22, 23). This fusion protein turns the WT1 tumour suppressor gene into a dominant oncogene by fusing the transcriptional activator at the N-terminus of EWS to the C-terminus DNA-binding site of WT1 (24). This fusion activates the same targets that WT1 would normally suppress.
Microscopically, DSRCT is composed of well-defined nests of small round blue tumour cells separated by abundant desmoplastic stroma (24). Immunohistochemically, DSRCT demonstrates a strikingly divergent differentiation. Typically, tumour cells are immunoreactive for epithelial (keratin and epithelial membrane antigen), mesenchymal (vimentin), myogenic (desmin), and neural (neuron-specific enolase and CD56) markers. Lae et al. (7) reported 32 tumours with the immunohistochemical features of DSRCT. Twenty-six tumours (81%) stained for desmin. Cytokeratin expression was demonstrated in 28 cases (88%) which stained for cytokeratin AE1/AE3. Twenty-seven tumours (84%) stained for NSE, and 7 of 30 tumours (23%) stained for CD99. Twenty-nine of 32 tumours (91%) stained for WT1 protein. In the present case, the tumour was positive for keratin AE1/AE3, EMA and desmin, and demonstrated nuclear positivity for polyclonal WT1. The unique expression of these multiple lineage antigens supported the diagnosis of DSRCT.
The extremely rare occurrence and young age of most patients with DSRCT make clinical treatment decisions difficult. Lal et al. (15) studied a cohort of 66 patients and found gross tumour resection to be highly significant in prolonging overall survival. The 3-year survival rate was 58% in patients who had undergone surgical resection, as compared to a 0% 3-year survival rate in non-resectable patients. Conversely, other authors have concluded that surgical excision does not significantly improve survival: Livaditi et al. (25) found that among those who underwent radical tumour excision with adjuvant chemotherapy, all had tumour recurrence within 2-6 months. The 3- and 5-year survival rates were 20% and 0%, respectively. Gil et al. (26) found no correlation between surgical excision and improved survival rate. A complete resection is rarely possible because the tumour tends to be large at presentation. Hassan et al. (27) reported that surgical resection prolonged survival in the patients. The median survival of patients who underwent complete surgical resection and chemotherapy was 34 months, whereas the median survival of patients who underwent chemotherapy alone was 14 months. Bertuzzi et al. (28) studied high-dose chemotherapy (ifosfamide, epirubicin and vincristine) in patients with various types of small round cell tumours. A subset with DSRCT had a very poor response to treatment (43% histological response versus 85% response in the other small round cell tumour types). Biswas et al. (29) reported a 39% response rate to multi-agent chemotherapy in a series of 18 patients. Lae et al. (7) reported 27 patients with DSRCT; out of them, 19 patients (70%) died of the disease and 8 patients (30%) were alive with evidence of active disease after a mean follow-up period of 26 months (range, 8-57 months).
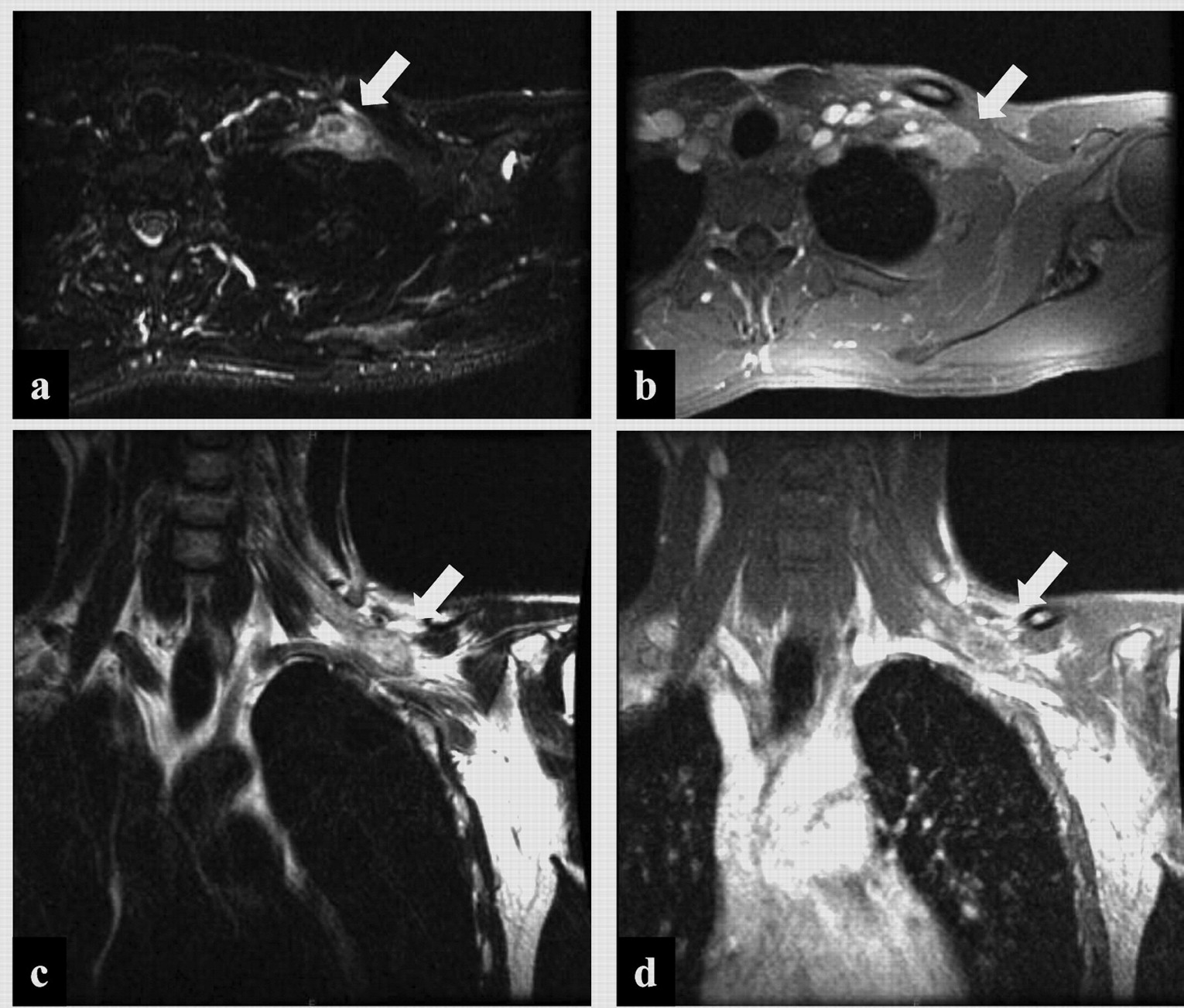
Magnetic resonance imaging. a: Axial T2-weighted image shows the tumor at the brachial plexus (arrow). b: Gadolinium-enhanced axial image shows ring enhancement (arrow). c: Coronal T2-weighted image shows the tumor in the left brachial plexus (arrow). d: Gadolinium-enhanced coronal image shows ring enhancement (arrow).

a: 201Tl scintigraphy revealed abnormal uptake at the left supraclaviclar fossa (arrow). b: 99mTc-MIBI scintigraphy revealed high accumulation at the lesion (arrow). c: FDG-PET scintigraphy shows very high accumulation at the brachial plexus (arrow).

Histology at biopsy. a: H&E section shows an architecture of variously sized, irregularly shaped nests of cells separated by a densely fibrotic and desmoplastic stroma, characteristic of DSRCT. b: Cytokeratin AE1, AE3 immunohistochemical stain shows staining in the tumor cells. c: Desmin immunohistochemical stain shows staining in the tumor cells. d: MIB-1 immunohistochemical stain shows partial staining in the tumor cells. e: FISH assay shows structural disruption of EWS gene.

The tumor disappeared in magnetic resonance imaging after the chemotherapy. a: Axial T2-weighted image shows the tumor at the brachial plexus. b: Gadolinium-enhanced axial image shows ring enhancement. c: Coronal T2-weighted image shows the tumor in the left brachial plexus. d: Gadolinium-enhanced coronal image shows ring enhancement.
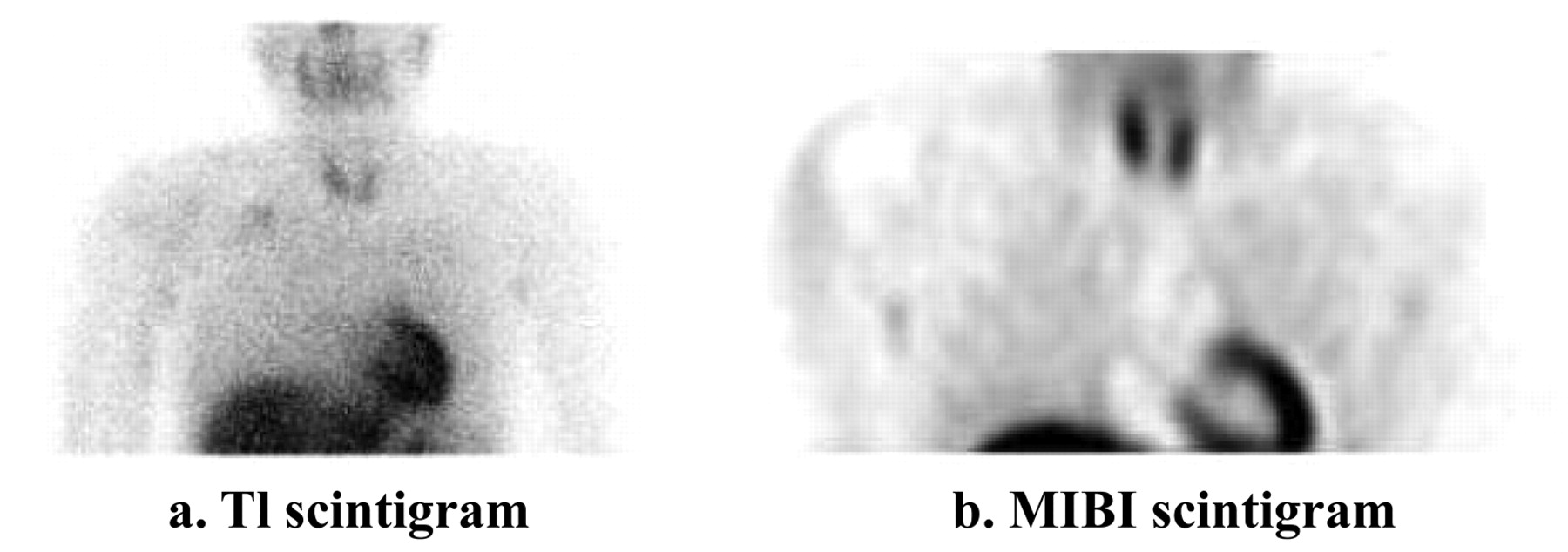
There is no uptake at 201Tl scintigraphy and 99mTc-MIBI scintigraphy after the chemotherapy. a: 201Tl scintigraphy. b. 99mTc-MIBI scintigraphy.
In the present case, caffeine-assisted chemotherapy was performed. Several studies have shown that caffeine dramatically enhances the tumouricidal effect of several antitumour drugs, such as cisplatin, thiotepa, doxorubicin, cyclophosphamide, mitomycin C, vincristine and methotrexate (30-33). Caffeine, which is a xanthine analogue, has a biochemical modulating effect as a DNA repair inhibitor and may inhibit postreplication repair of sublethally damaged DNA (34). The proposed mechanisms of the enhanced antitumour effect of caffeine are to induce G1/S-arrest and to reverse or abrogate the G1/S and the G2/M checkpoint delay periods (35), or to initiate rapid apoptosis in spindle checkpoint-arrested cells. At this checkpoint, p21-activated kinase 1 (PAK1) is at least a significant contributor to the caffeine-induced apoptosis in response to either microtubule poisons or DNA-damage (36). Furthermore, Bode and Dong (35) reported that the effect of caffeine on cell cycle seems to depend on its concentration. In the host institute, caffeine-assisted chemotherapy induced a complete response in more than 80% of patients with osteosarcoma (37). A high rate of clinical response to caffeine-assisted chemotherapy was also observed in patients with high-grade soft-tissue sarcoma/metastatic carcinoma and lymphoma of bone and soft tissue (38, 39). The present case showed good response to caffeine-assisted chemotherapy. Currently, there has been no recurrence and no functional disorder has occurred in the patient's upper limb 39 months after the chemotherapy.
In summary, despite its rarity, DSRCT should be a differential diagnosis of malignant small round cell tumour at any site. Although the current treatments for DSRCT are not curative, caffeine-assisted chemotherapy combined with radiotherapy was effective for DSRCT in the present case. It is therefore concluded that caffeine-assisted chemotherapy should be one of the treatment options for DSRCT.
- Received April 22, 2010.
- Revision received May 25, 2010.
- Accepted June 3, 2010.
- Copyright© 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}