


Abstract
Gingival squamous cell carcinoma is a rare form of cancer that accounts for less than 10% of all head and neck cancers. Targeted therapies with natural compounds are of interest because they possess high efficacy with fewer side-effects. Methylsulfonylmethane (MSM) is an organic sulfur-containing compound with anticancer activities. The main goal of this study was to induce proliferation inhibition and apoptosis in the metastatic YD-38 cell line. MSM up-regulated expression of P21Waf1/Cip1 and P27Kip1 genes and down-regulated expression of cyclin D1 (CCND1) and CDK4. Moreover, treatment with MSM induced apoptosis and up-regulation of BAX in YD-38 cells. In accordance, the expression of the BCL-2 and BCL-XL, were inhibited, indicating the role of mitochondria in MSM-induced apoptosis. Analysis of mitochondrial integrity showed a loss of mitochondrial potential with an increased level of cytochrome c in the cytosol compared to mitochondria. Active CASPASE-3 (CASP3) was also observed, confirming that MSM-induced apoptosis is caspase-mediated.
Gingival squamous cell carcinomas (GSCCs) are rare cancers, accounting for less than 10% of all head and neck cancers (1). GSCC has a high risk of metastasis and consequent death with bone invasion of high-grade tumors (2). Recent advances in chemo, radiation, and surgical therapies offer near-complete recovery for patients with oral malignancies. Modulating the expression and function of genes and impairing their roles in numerous tumor-dependent signaling pathways by specifically targeting them using natural compounds has become a new line of therapy. This targeted therapy holds advantages because it can reduce treatment-induced adverse effects without compromising the therapeutic efficiency and efficacy.
Cancer treatment mainly depends on the ability of the anticancer drug to inhibit cell cycle progression and induce apoptosis. Control of cell-cycle progression is the beginning of these processes. Cyclin-dependent kinases (CDKs) and activated cyclin D1 (CCND1) play a major role in cell-cycle progression. CDK inhibitors regulate the activity of CDKs/CYCLINs (3). Cyclin E (CCNE1) also participates in cell-cycle progression. The tumor suppressors cyclin dependent kinase inhibitor 1A (CDKN1A, also known as P21WAF1/Cip1) and cyclin dependent kinase inhibitor 1B (CDKN1B, also known as P27Kip1) prevent phosphorylation of the targets of D/E-type cyclins (4). P21WAF1/Cip1 mediates P53-dependent G1 growth arrest (5) and suppresses tumor growth by inducing cell-cycle arrest and also acts as a key factor in multiple tumor suppressor pathways that regulate cell death. P27Kip1 acts as a tumor suppressor protein by regulating the transition from G0 to S phase during the cell cycle by CDKs. Down-regulation of P27Kip1 expression during G1 phase allows the transcriptional activation of genes required for G1/S transition (6).
Apoptosis involves changes in cellular morphology such as membrane blebbing, cell shrinkage, chromatin condensation, nuclear fragmentation and DNA fragmentation (7, 8). The progression of cancer depends on the loss of control over the induction of apoptosis (9). Intrinsic apoptosis requires Bcl-2, Bax, and mitochondrial signals, whereas extrinsic apoptosis requires signaling from death receptors in the plasma membrane. Caspases are the major effectors that play an important role in apoptosis (10).
RT-PCR primer sequences, annealing temperature and respective product sizes.
Treatment of cancer with natural compounds is a good therapeutic approach in order to avoid side-effects of drugs. Methylsulfonylmethane (MSM) is a natural organic sulfur-containing compound, which is also known as dimethyl sulfone. It is mainly present in foods like fruits, vegetables, and beverages. MSM can be taken up through the diet (11, 12). MSM is used as a drug to alleviate pain and improve physical function in osteoarthritis (13). MSM has anti-cancer activities such as wound healing and contact inhibition, and can inhibit cell migration through the extracellular matrix in various types of cancers (14, 15). Our recent study proved that MSM is able to up-regulate the expression levels of growth hormone receptor (GHR) and signal transducer and activator of transcription 5b (STAT5b) in osteoblast-like cells (16). It is also able to inhibit in vitro ketosis in mouse hepatocytes (17).
In the current study, we assessed the effect of MSM on gingival squamous cell adenocarcinoma. We hypothesized that MSM would be able to up-regulate the expression levels of P21WAF1/Cip1, P27Kip1 and BAX and thereby arrest cell cycle progression and induce mitochondrial apoptosis in gingival cancer cells.
Materials and Methods
Antibodies and reagents. RPMI-1640 medium, 10% fetal bovine serum (FBS), and 0.05% trypsin–ethylenediaminetetraacetic acid were purchased from Gibco-BRL (Grand Island, NY, USA). BAX, BCL-2, BCL-XL, CASPASE-3, β-actin antibodies and secondary antibodies (rabbit and goat anti-mouse IgG–horse radish peroxidase) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). An enhanced chemiluminescence detection kit (ECL plus), RT-PCR Premix kits, oligo d(T), and BCL-2, BCL-XL, BAX and 18S primers for RT-PCR were purchased from Bioneer (Daejeon, Korea). DiOC6 was obtained from Sigma (St. Louis, MO, USA). A mitochondria isolation kit and a Coomassie (Bradford) Protein Assay Kit were purchased from Thermo Scientific (Waltham, MA, USA). Restore Western Blot Stripping Buffer and NE-PER kits were purchased from Pierce (Rockford, IL, USA). An RNeasy mini kit and Qiaprep spin mini prep kits were purchased from Qiagen (Hilden, Germany). MSM was purchased from Fluka/Sigma Co. (St. Louis, MO, USA).
Cell culture and treatment. The gingival cancer cell line YD-38 was maintained in RPMI-1640 containing 10% FBS and 1% penicillin/streptavidin at 37°C in 5% CO2. Cells were placed in airtight chambers (Nu Aire, Plymouth, MN, USA). At the beginning of each experiment, cells were re-suspended in the medium at a density of 2.5×105 cells/ml. Cells were treated with 200 mM MSM.
Cell proliferation inhibition. Cell viability was assayed by measuring blue formazan that was metabolized from 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetra-zolium bromide (MTT) by mitochondrial dehydrogenase, which is active only in live cells. Cells were re-suspended in the medium one day before drug treatment at 3×103 cells per well of 96-well culture plates. Liquid medium was replaced with a fresh medium for control (vehicle). Cells were incubated with various concentrations of MSM: MTT (5 mg/ml) was added to each well and incubated for 4 h at 37°C. The formazan product was dissolved by adding 200 μl dimethyl sulfoxide (DMSO) to each well, and the absorbance was measured at 550 nm on an Ultra Multifunctional Microplate Reader (TECAN, Durham, NC, USA). All measurements were performed in triplicate, and experiments were repeated at least three times.
Western blotting. Cells were treated with MSM (200 mM) for different time periods. Cells were lysed on ice with radioimmunoprecipitation (RIPA) lysis buffer containing 1X BD Baculogold protease inhibitor cocktail (BD Biosciences, CA, USA) and 1X PhosSTOP phosphatase inhibitors (Roche, USA). Cells were disrupted by aspiration through a 23-gauge needle and centrifuged at 15,000 rpm for 10 min at 4°C to remove debris. Protein concentrations were measured using the Bradford method. Equal amounts of total protein were resolved by SDS-PAGE and transferred onto a nitrocellulose membrane. Blots were blocked for 1 h with 5% skim milk. The membranes were probed with primary antibodies overnight at 4°C, followed by HRP-conjugated secondary antibodies. Detection was performed using the ECL plus detection kit and LAS-4000 imaging device (Fujifilm, Japan). Quantification analysis were done by using multigauge software.
Reverse transcription polymerase chain reaction (RT-PCR). Cells were treated with MSM (200 mM) for determined times. Total RNA was extracted with an RNeasy Mini Kit and quantified spectrophotometrically at 260 nm. RT-PCR analysis for P21WAF1/Cip1, P27Kip1, CDK4, CCND1, BAX, BCL-2, BCL-XL, and 18S RNA was performed. cDNA was synthesized from total RNA by reverse transcription at 42°C for 1 h and 80°C for 15 min using a first strand cDNA synthesis kit (Bioneer, Korea). PCR was conducted with cDNA as a template for 25-30 cycles of denaturation at 94-95°C, annealing at 56-60°C, and extension at 72°C. The primers used for the amplification are listed in the Table I. PCR products were analyzed by 1% agarose gel electrophoresis followed by staining with ethidium bromide. Quantification analysis was performing by using multigauge software.
Cell-cycle analysis. The DNA content of treated and untreated cells was determined with a BD Cycletest Plus DNA reagent kit (BD Biosciences, CA, USA) following the manufacturer's protocol. Briefly, approximately 5×105 cells, induced or not induced with MSM for indicated periods of time, were separated, washed twice with PBS and permeabilized using trypsin buffer. The RNA interaction with propidium iodide (PI) was neutralized by treating the cells with trypsin inhibitor and RNase buffer. The samples were then stained with PI for 30 min in the dark and analyzed using a FACS Calibur flow cytometer (BD Biosciences, SanJose, CA, USA).
Apoptosis analysis. Fluorescein-conjugated annexin V (annexin V-FITC) was used to quantify the percentage of cells undergoing apoptosis. Drug-treated cells were washed, resuspended in binding buffer at a concentration of 1×106 cells and stained with annexin V-FITC and propidium iodide. After incubation for 15 min at room temperature in the dark, the percentage of apoptotic cells was analyzed using flow cytometry on FACS calibur. Camptothecin (10 μM) was used as a positive control. Percentage cell count shown in Table II. Analysis were done by using Cyflogic software.
Apoptotic DNA ladder analysis. Cells were treated with 200 mM MSM for 24 h, collected by centrifugation, and DNA ladder analyses were carried out using Apoptotic DNA ladder kit (Roche, Basel, Switzerland). DNA was isolated as per kit protocol and products were then analyzed by electrophoresis in 1% agarose gel containing ethidium bromide.
Isolation of mitochondria. Mitochondria from MSM-treated and untreated cells were isolated using a mitochondria isolation kit following the manufacturer's protocol. Cells (2×107) were seeded and grown to 80% confluence. Cells were then collected by centrifugation, treated with mitochondria isolation reagent and incubated on ice. Following incubation, Reagents B and C were added with mixing and incubation on ice between each addition. The mixture was centrifuged at 700 × g for 10 min; the supernatant was collected and centrifuged again. Then the supernatant (cytosol) and the mitochondrial pellet were washed with Reagent C and used for downstream applications.
Percentage cell count obtained after the analysis with the Cyflogic software.
Measurement of mitochondrial membrane potential (ΔΨm). Changes of mitochondrial membrane potential (ΔΨm) were measured by the DiOC6 staining method. Briefly, cells treated or not with MSM or camptothecin (positive control) were washed and suspended in 0.1 μM DiOC6 solution. Cells were then incubated at 37°C for 20 min and washed with pre-warmed PBS. Cells were then analyzed using FACS Calibur.
Statistical analysis. All experiments were repeated three times and the results were expressed as mean±SEM. Statistical analysis was done with Student's t-test or ANOVA test of the SAS program.
Results
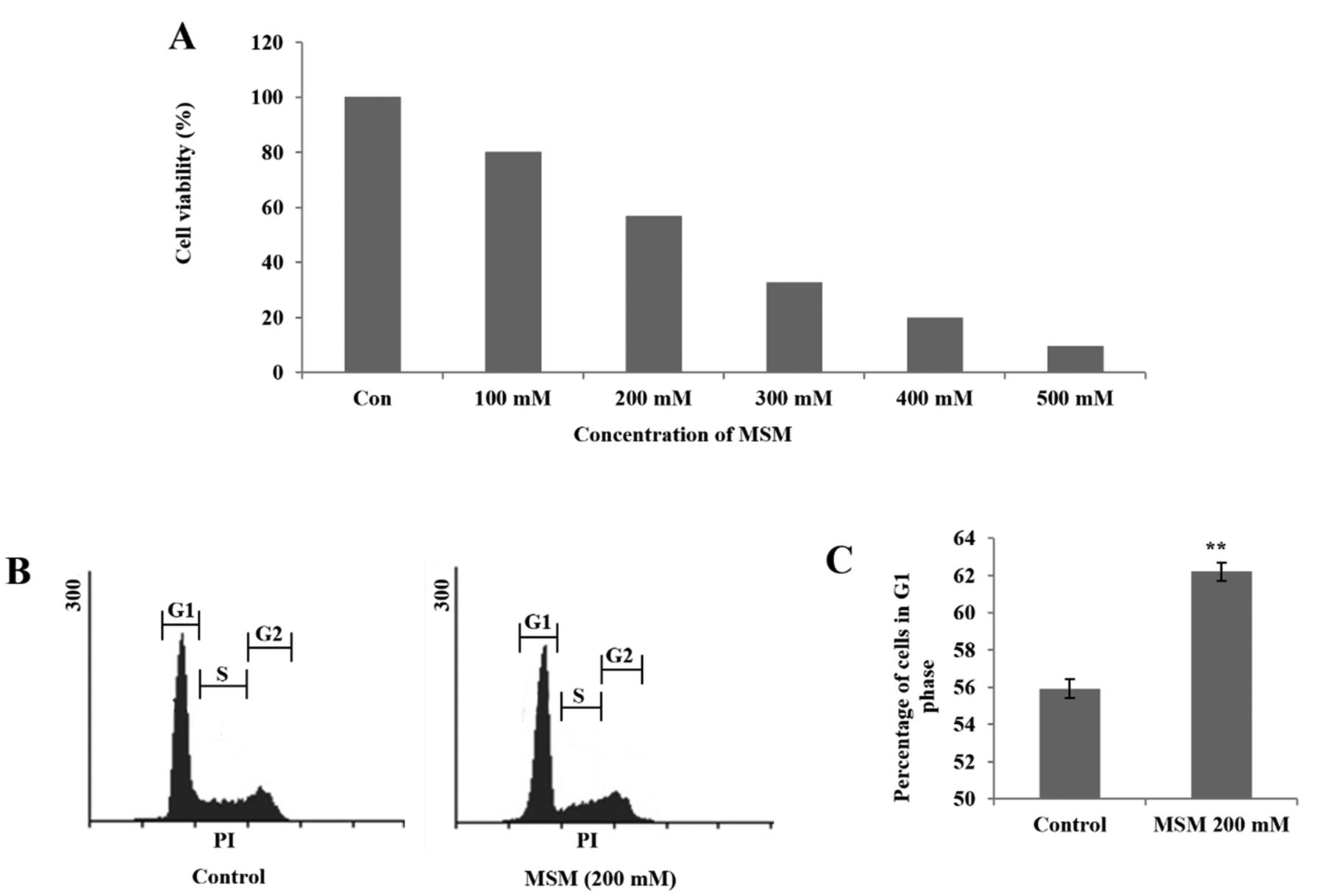
MSM inhibits cell viability in a concentration-dependent manner and induces G1 arrest in gingival cancer cells. To determine whether MSM inhibits proliferation of a gingival squamous adenocarcinoma cell line, the number of treated cells during the logarithmic phase was compared with the number of non-treated control cells. Cell growth was inhibited by approximately 43% after 24 h of treatment with 200 mM MSM (Figure 1A). This concentration was considered to be the IC50 and was used for further experiments. During the treatment period of 24 h, the cells may undergo apoptosis. Therefore, to analyze cell cycle arrest, we treated cells with 200 mM MSM for 12 h. After treatment, cells were stained with propidium iodide (PI) and the distribution of nuclei was analyzed. The results indicated that MSM treatment induced the accumulation of cells in G1 phase in comparison with untreated control cells (Figure 1B). The graphical representation showed G1 phase cell cycle arrest in MSM-treated cells (Figure 1C).

MSM inhibits cell viability and induces G1 arrest in gingival cancer cells. (A) Inhibition of the proliferation of YD-38 gingival cancer cells by MSM in a dose-dependent manner in a 24-h treatment period. (B) Histogram showing G1 arrest after treatment with 200 mM MSM for 12 h in YD-38 cells. (C) Graphical representation of the cell distribution in G1 phase. Experiments were repeated three times, and mean values are presented in the final graph. Statistical analysis was performed using the Student's t-test (**p<0.01).
MSM increases the expression of P21 and P27 and inhibits the expression of cyclin complexes and CDK4. The expression of genes that might be responsible for the cell cycle arrest, was analyzed. Inhibition of the G1/S phase transition depends primarily on the levels of p21Waf1/Cip1 and p27Kip1, and their loss results in uncontrolled cell proliferation. Figure 2A shows RT-PCR analysis of cells treated with 150 and 200 mM MSM along with untreated control cells. Expression of both p21Waf1/Cip1 and p27Kip1 was up-regulated upon MSM treatment, indicating cell cycle arrest, while expression of CCND1 and CDK4 was down-regulated, confirming G1 arrest. Transcription level analysis showed a significant and concomitant level of regulation upon MSM treatment (Figure 2B). To confirm the effect of MSM on cell cycle, we analyzed its effect on the levels of proteins responsible for cell cycle progression. Figure 2C shows western blotting analysis of MSM-treated and untreated cells. MSM reduced the levels of CCND1, CCNE1, and CDK4 in a concentration-dependent manner. This result confirmed the MSM-induced G1 arrest in this cell line. Graphical representation of the levels of CCND1 and CCNE1 showed significant inhibition by 200 mM MSM in comparison with untreated control cells (Figure 2D).
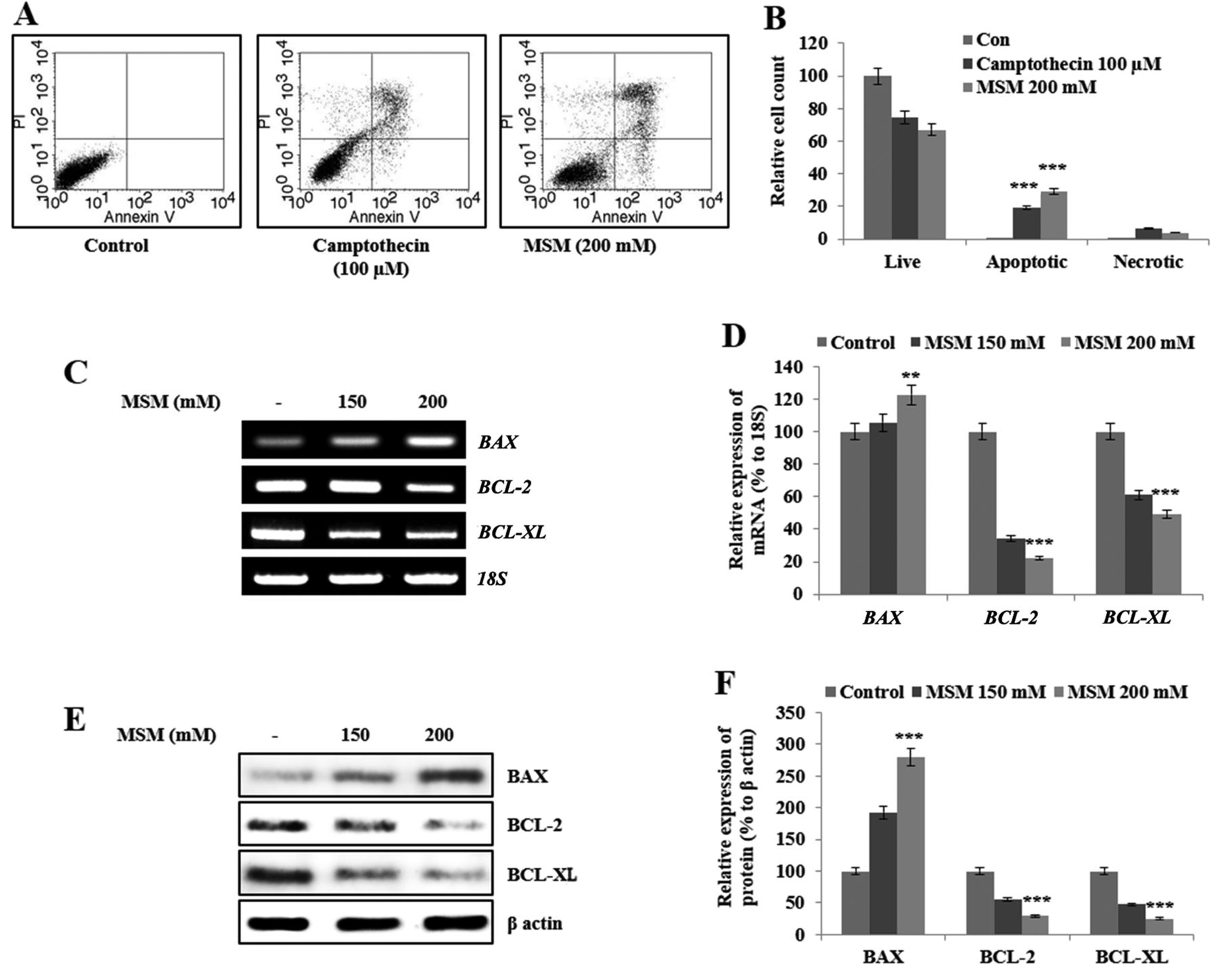
MSM induces apoptosis in gingival cancer cells by down-regulating expression of the anti-apoptotic genes Bcl-2 and Bcl-xL and up-regulating expression of Bax. Our next aim was to assess the effect of MSM on cell death. We observed cell cycle arrest after 12 h of MSM treatment; therefore, we analyzed apoptosis after 24 h of MSM treatment. The representative number of apoptotic cells were much higher after MSM treatment compared to un-treated cells or to cells treated with camptothecin (100 μM), used as a positive control (Figure 3A). Graphical representation of apoptosis analysis showed approximately 30% increase in the relative cell count of apoptotic cells by MSM treatment compared to control cells (Figure 3B). We analyzed the levels of anti-apoptotic proteins and their transcripts after treatment with 150 or 200 mM MSM. The results of RT-PCR analysis showed elevated expression of BAX and decreased expression of BCL-2 and BCL-XL in treated cells in comparison with the control (Figure 3C). Graphical analysis of genes responsible for apoptosis showed that these changes in expression were significant (Figure 3D). The MSM-induced changes in the expression of these genes are consistent with the induction of apoptosis. To confirm the results of RT-PCR analysis, we examined the protein levels by western blotting. The expression pattern was similar to that seen at the transcriptional level; BCL-2 and BCL-XL were down-regulated, whereas BAX was up-regulated by MSM treatment (Figure 3E). Graphical analysis showed that the MSM-induced changes in the levels of BCL-2, BCL-XL, and BAX were significant (Figure 3F).

MSM regulates the expression of p21Waf1/Cip1, p27Kip1, cyclin complexes, and CDK4. (A) RT-PCR analysis of control cells and cells treated with 150 or 200 mM MSM for 12 h. (B) Graphical representation of the transcriptional levels of P21Waf1/Cip1, P27Kip1, CCND1, and CDK4 after MSM treatment. (C) Western blot analysis of control cells and cells treated with 150 or 200 mM MSM for 12 h. (D) Graphical representation of the levels of CCND1, CCNE1 and CDK4 after MSM treatment for 12 h. Statistical analysis was performed using ANOVA-test (**p<0.01; ***p<0.001).
MSM regulates the levels of mitochondrial BCL-2, BCL-XL, and BAX and induces loss of mitochondrial trans-membrane potential (MTP) in gingival cancer cells. To check the effect of MSM on mitochondrial apoptosis, we isolated mitochondrial and cytosolic fractions after treatment of cells with MSM and examined the mitochondrial and cytosolic levels of BCL-2, BCL-XL, and BAX. In MSM-treated cells, the level of mitochondrial BAX was elevated, whereas the levels of mitochondrial BCL-2 and BCL-XL were lower than in untreated cells (Figure 4A). This indicates an increased mitochondrial localization of BAX. Western blotting analysis of the cytosolic fractions showed similar patterns. Levels of BCL-2 and BCL-XL were decreased in whole cell lysates. To evaluate mitochondrial trans-membrane potential (MTP) after MSM treatment, we stained treated and untreated cells with DiOC6. MTP was lower in MSM-treated cells than in control cells. In Figure 4B, region R1 denotes the MTP loss. The number of cells present in the R1 region was higher upon MSM treatment than in the control. The loss of MTP was statistically significant (Figure 4C).
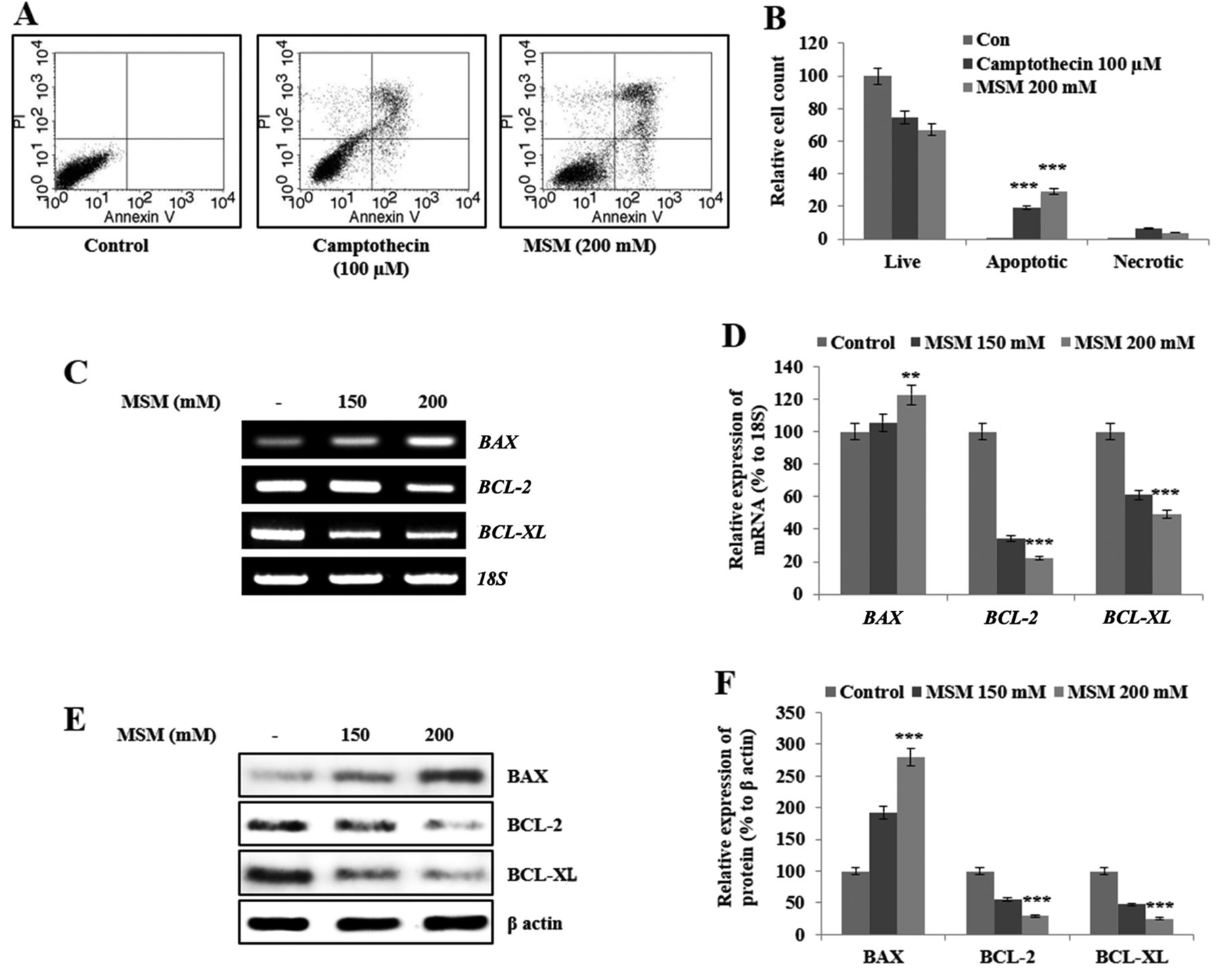
MSM induces apoptosis by regulating the expression of BCL-2, BCL-XL, and BAX. (A) Annexin V FITC vs. PI staining showed apoptosis induction. (B) Graphical analysis of the percentage of apoptotic cells upon control, camptothecin, and MSM treatment. Statistical analysis was performed using the Student's t-test (***p<0.001). (C) RT-PCR analysis of anti-apoptotic genes after treatment with 150 and 200 mM MSM for 24 h. (D) Graphical representation of the levels of BAX and anti-apoptotic protein transcripts after MSM treatment for 24 h. (E) Western blot analysis of protein levels after treatment with MSM for 24 h. (F) Graphical representation of the levels of BAX, BCL-2, and BCL-XL after MSM treatment. Data are representative of three independent experiments. Statistical analysis was performed using ANOVA-test (**p<0.01; ***p<0.001).
MSM induces the release of cytochrome c (CYC) into the cytoplasm in gingival cancer cells. The induction of mitochondrial apoptosis depends on the release of cytochrome C (CYC) from the mitochondria into the cytosol. To evaluate the CYC release, cells were treated with MSM and the presence of CYC in the whole cell lysates as well as the mitochondrial and cytosolic fractions of treated and untreated cells were examined. The level of CYC in whole-cell lysates was increased upon MSM treatment, which indicates the release of CYC from the mitochondria (Figure5A). To confirm this, we also checked the localization and release of CYC into the cytosol. Analysis of CYC in the mitochondria showed its level was reduced after MSM treatment in comparison with control cells, whereas cytosolic fractions showed an elevated level of CYC after MSM treatment (Figure 5B). This clearly proved the release of CYC from the mitochondria into the cytosol.

MSM regulates the levels of mitochondrial apoptotic factors and induces loss of MTP. (A) Western blot analysis of the protein levels of mitochondrial and cytosolic BCL-2, BCL-XL, and BAX after MSM treatment for 24 h. (B) Analysis of the loss of MTP using DiOC6 in YD-38 cells after treatment with 200 mM MSM for 24 h. (C) Graphical representation of the percentage loss of MTP. Statistical analysis was performed using the Student's t-test (***p<0.001).
MSM induces caspase-dependent apoptosis and DNA fragmentation in gingival cancer cells. Active (cleaved) caspases are needed for mitochondrial apoptosis. Analysis of whole-cell lysates from MSM-treated and untreated cells showed the presence of cleaved CASPASE-3 after MSM treatment, which confirmed the induction of apoptosis and also indicated that caspases play a major role in apoptosis induced by MSM (Figure 6A). We examined DNA fragmentation to confirm the ability of MSM to induce apoptosis. DNA ladder formation was observed in MSM-treated cells, and the intensity of the ladder increased in a concentration-dependent manner (Figure 6B). These results confirmed the ability of MSM to induce apoptosis in gingival cancer cells.
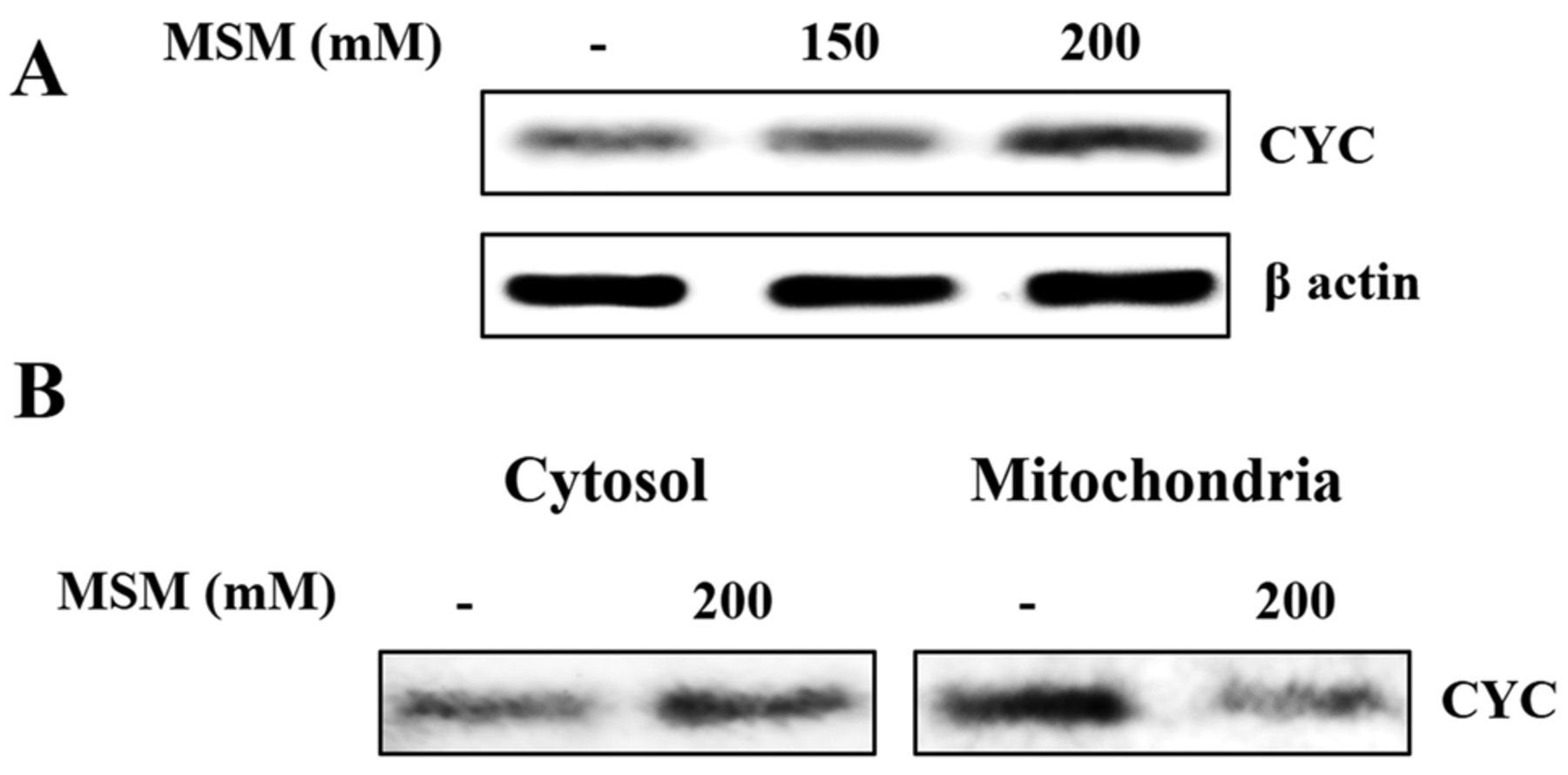
MSM-induced release of CYC into the cytoplasm. (A) Western blot analysis of CYC in a whole cell lysate upon treatment with 200 mM MSM for 24 h. (B) Western blot analysis of mitochondrial and cytosolic fractions showing the release of CYC into the cytosol.
Discussion
Treatment of oral cancer remains a challenge, despite development of a variety of drugs against various types of cancers. Because most of the chemical drugs have many side-effects, even if they help patients recover from cancer, they cause some other diseases that may be life-threatening. The use of natural compounds for cancer treatment started years ago. The main advantage of natural compounds is that they have less severe side effects than anti-cancer drugs. MSM is a natural organic sulfur-containing compound with anti-cancer activities. Some side-effects of MSM have been detected so far. MSM has many anticancer activities such as cell-cycle arrest and induction of apoptosis, inhibition of angiogenesis, inhibition of migration and invasion, and thereby inhibition of metastasis (12). Here, we analyzed the effect of MSM in gingival squamous adenocarcinoma cells. To determine the IC50 of MSM in gingival cancer cells, we checked which concentration induces apoptosis after 24 h of treatment. A MSM concentration of 200 mM resulted in almost 43% cell death compared with the control. We used 200 mM MSM for further analysis.
MSM treatment for 24 h may induce apoptosis; therefore, we performed treatment for 12 h to check the ability of MSM to induce cell cycle arrest. In mammalian cells, proliferation is mainly controlled in G1 phase and this affects the other phases (18). We observed an arrest in G1 phase (Figure 2A) in MSM-treated cells compared with control cells. This supported our assumption about the ability of MSM to induce cell-cycle arrest after 12 h of treatment. To confirm this, we analyzed the genes and proteins that might mediate this effect of MSM. Cell cycle arrest occurs mainly due to DNA damage caused by drugs which stabilizes P53 protein. This process is mediated by a cyclin kinase inhibitor and the cell cycle regulators p21Waf1/Cip1 and p27Kip1, which are up-regulated during cell cycle arrest. CDKs are activated by binding to cyclin complexes, and their activity is inhibited by cyclin kinase inhibitors (19). Thus, CDKs and cyclin complexes should be down-regulated by expression of CDK inhibitors when cell cycle arrest happens. This study showed down-regulation of CCND1, CCNE1 and CDK4, and up-regulation of p21Waf1/Cip1 and p27Kip1 at both the transcriptional and translational levels upon MSM treatment, which demonstrates its anti-cancer activity. By contrast, P21Waf1/Cip1 and P27Kip1 are considered to be tumor suppressor genes, i.e., up-regulation of these genes suppresses tumor growth. Thus, the up-regulation of p21Waf1/Cip1 and p27Kip1 may also account for the anti-cancer activity of MSM.

MSM induces caspase-dependent apoptosis and DNA fragmentation. (A) Western blot analysis of a whole cell lysate showing CASPASE-3 activation after treatment with 200 mM MSM for 24 h. (B) MSM-induced DNA fragmentation in YD-38 cells. Formation of DNA fragments after treatment with 150 or 200 mM MSM for 24 h was assessed with an apoptotic DNA ladder kit.
Normally, after cell-cycle arrest, cells tend to die. After treatment with 200 mM MSM for 24 h, 40% of cells underwent apoptosis with respect to untreated control cells. This preliminary result was confirmed by the formation of a DNA ladder. After cell-cycle arrest, the anti-apoptotic members of the Bcl-2 family determine whether the cell proceeds to apoptosis (20, 21). Mitochondria have a crucial function in apoptosis (22-24). In the mitochondria, Bcl-2 and Bcl-xL act as anti-pore factors; they inhibit the release of CYC into the cytosol and inhibit apoptosis. MSM inhibited the expression of BCL-2 and BCL-XL at both the transcriptional and translational levels, in line with the ability of MSM to induce apoptosis. BAX is a pro-apoptotic protein that acts as a pore factor in the mitochondria and also plays an important role in apoptosis (25). The expression of BAX is elevated during apoptosis. BAX determines whether the cell proceeds to mitochondrial apoptosis (26, 27).
In the intrinsic apoptotic pathway, cell death is mainly mediated through mitochondria (28). When a pro-apoptotic stimulus is perceived by the cell, the expression of anti-apoptotic factors like BCL-2 decreases and that of pro-apoptotic factors like BAX increases. This triggers mitochondrial apoptosis by opening the pores of mitochondria, which leads to the loss of mitochondrial membrane integrity. A loss of ATP also occurs inside the mitochondria. The loss of MTP is a key factor in mitochondrial apoptosis, which occurs due to the activity of the membrane form of BAX. Mitochondrial pore and anti-pore factors (BAX and BCL-2, respectively) play a role in the maintenance of MTP (29, 30). As a result of MTP loss, the membrane is opened by BAX and this leads to the release of CYC from the mitochondria into the cytosol. In the cytosol, CYC helps promote caspase-mediated apoptosis. Caspases are the effector enzymes in apoptosis.
An increase in the level of CYC was observed in total cell lysates after MSM treatment, which indicates mitochondrial apoptosis. To confirm this, we checked the levels of CYC in the mitochondria and cytosol, and the results were fascinating. Upon MSM treatment, the level of CYC in the mitochondria decreased and that in the cytosol increased, indicating a release of CYC from the mitochondria into the cytosol. As a result of CYC release, caspase protein is activated; indeed, its expression level was also up-regulated upon MSM treatment, which confirms the induction of apoptosis through the caspase-dependent pathway. Among all caspases, CASPASE-3 is a major enzyme that participates in the intrinsic pathway of apoptosis. The analysis of active CASPASE-3 confirmed the induction of apoptosis by treatment with 200 mM MSM.
In conclusion, this study demonstrated the use of a natural compound to induce cell-cycle arrest and mitochondrial apoptosis. MSM has various anti-cancer activities. The present study indicates that it induces cell cycle arrest through the p21Waf1/Cip1-dependent pathway and induces mitochondrial apoptosis through CYC and the caspase-dependent pathway. The added advantage of MSM may be that it is non-toxic and has no side effects. These findings suggest that MSM is a good anti-cancer drug in gingival squamous adenocarcinoma.
Acknowledgements
This study was supported by the Konkuk University, Seoul, Republic of Korea in 2015.
Footnotes
↵* These Authors contributed equally to this study.
This article is freely accessible online.
- Received January 20, 2017.
- Revision received March 1, 2017.
- Accepted March 2, 2017.
- Copyright© 2017, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}