


Abstract
The leading cause of lung cancer is exposure to cigarette smoke and other environmental pollutants, which include formaldehyde, acrolein, benzene, dioxin, and polycyclic aromatic hydrocarbons (PAHs). PAHs and dioxins are exogenous ligands that directly bind to the aryl hydrocarbon receptor (AhR), a transcription factor that activates xenobiotic metabolism, histone modification (an important step in DNA methylation) and, ultimately, tumorigenesis. In this review article we summarize the current understanding of AhR and its role in the development of lung cancer, including its influence on cell proliferation, angiogenesis, inflammation, and apoptosis.
- Aryl hydrocarbon receptor
- lung cancer
- tumorigenesis
- dioxin
- polycyclic aromatic hydrocarbons
- cigarette smoke
- review
Lung cancer is the leading cause of cancer-related death worldwide, accounting for nearly one third of all cancer deaths (1). Lung cancer has only a 15% 5-year survival rate because most patients present with advanced disease. When lung cancer is detected at an early stage, surgical resection results in a higher cure rate. Understanding the early molecular events in lung carcinogenesis may lead to the discovery of new markers for early detection or targets for therapy. Exposure to cigarette smoke and environmental pollutants is the major culprit in the development of lung cancer. Individuals who have smoked are 10- to 20-times more likely to develop lung cancer than never-smokers. Other risk factors include exposure to second-hand smoke, radon and asbestos, having a first-degree relation with lung cancer, and a diagnosis of chronic obstructive pulmonary disease (COPD)/emphysema.
The most commonly proposed mechanisms for lung carcinogenesis are chronic inflammation and defects in the DNA repair capacity in the epithelial cells of the central and small airways. The fine particles in cigarette smoke and air pollution as well as volatile chemicals including formaldehyde, acrolein, benzene, polycyclic aromatic hydrocarbons (PAHs) and N-nitrosamines, are deposited on the stem cell-like distal bronchoalveolar epithelial cells, and can alter genomic pathways. Many of these carcinogenic chemicals exert direct biological effects by binding to a cytosolic receptor, the aryl hydrocarbon receptor (AhR). Activation of AhR has a variety of downstream effects which influence tumorigenesis, inflammation, formation of DNA adducts, cell proliferation, and loss of cell–cell adhesion. Therefore, AhR likely plays an important role in cigarette smoke-induced lung cancer. Understanding the role of AhR in lung tumorigenesis may lead to the identification of improved markers for early diagnosis and prognosis of lung cancer, and novel targets for therapy. In this review, we summarize the current knowledge of the AhR and its role in pulmonary carcinogenesis.
Aryl Hydrocarbon Receptor
The aryl hydrocarbon locus, which includes AhR, AhR nuclear translocator (ARNT), and AhR repressor (AHRR), was first identified in 1972 (2). AhR is induced by PAHs and its primary role is the control of xenobiotic metabolism through cytochrome P450. In 1982, Poland and Knutson showed that 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and components of cigarette smoke, like benzo(a)pyrene (BaP), and PAH compounds, exert their biological effects by binding directly to a cytosolic receptor, now known as the AhR (3). The AhR is a member of the family of basic helix-loop-helix/Per-ARNT-Sim (bHLH-PAS) transcription factors (Figure 1). Other members of this family include ARNT and AHRR. In its inactivated state, AhR forms a complex with heat-shock protein HSP90, X-associated protein-2 (XAP2, AIP, or ARA9) and a co-chaperone protein p23 (4). When exogenous ligands bind and activate AhR, AhR dissociates from this complex and translocates into the nucleus after it dimerizes with ARNT to form a complex that is able to bind to xenobiotic response elements (XREs) of AhR-responsive genes. In response to hypoxia, ARNT can serve as an obligate hetero-dimerization partner for hypoxia-inducible-factor-1α (HIF-1α) and regulate vascular endothelial growth factor (VEGF) signaling, a key element for angiogenesis (5). AHRR, on the other hand, represses AhR transcriptional activity by competing with ARNT dimerization and subsequent binding to an XRE sequence. AHRR expression is induced by the AhR/ARNT dimer binding to XREs located in the 5’-flanking regions of the AHRR gene. Therefore, AhR and AHRR, together with ARNT, form a regulatory loop in which AhR/ARNT activates the AHRR gene, while AHRR/ARNT inhibits the function of AhR (6).
AhR is expressed in all tissues and is highly expressed in liver, adipose tissue, and bronchial epithelial cells. Because of its high level of expression in human bronchial epithelial cells, AhR has many physiological consequences in the lung through its effects on cell proliferation and differentiation, cell-cell adhesion interaction, cytokine expression, mucin production, and xenobiotic metabolism (7, 8). For example, AhR plays a role in chronic bronchitis, mediating the increased production of mucin in a clara-cell-derived cell line (9). The receptor plays a significant role in asthma and COPD through regulation of regulatory T-cell (T-reg) and T-helper-17 cell (Th17) cell differentiation (10). Recent studies have shown that it also plays an important role in the development of lung cancer.
Cross-Talk with other Pathways
XRE: Cytochrome P450. As mentioned, the best-understood function of AhR is its role in xenobiotic metabolism and induction of the cytochrome P450 enzymes. AhR dimerizes with ARNT and binds to xenobiotic response elements which regulate the expression of cytochrome P450. Cytochrome P450 is a group of enzymes that are key players in phase-I and phase-II enzyme reactions responsible for metabolizing carcinogens to their inactive derivatives. Phase-I-dependent oxidation of xenobiotics is important in the detoxification of carcinogens mainly through a mono-oxygenation of organic substances. Phase-II-dependent reactions further conjugate phase-I-modified compounds to allow their subsequent excretory (11). However, this step can rarely produce a more carcinogenic product than was the initial compound. In in vivo models, AhR has been shown to cause induction of cytochrome P450, family-1, member-A1 (CYP1A1) and CYP1B1 in human lung tissues after exposure to cigarette smoke (12). BaP, a constituent of cigarette smoke and a potent carcinogen, activates the AhR transcription factor, and increases phase I xenobiotic metabolism, including CYP1A1, CYP1B1, and other enzymes (12-14). AhR and its close relationship with cytochrome P450 play an important role in cigarette smoke-induced diseases, including lung cancer.
Nuclear factor kappa light-chain-enhancer of activated B-cells (NF-κB). NF-κB is a family of transcription factors that regulate many aspects of cellular physiology, such as inflammation, cell survival, and proliferation. The family includes subunits of NF-κB1 (p50), NF-κB2 (p52), p65 (RelA), RelB, and c-Rel. Dimerization of NF-κB subunits is essential for transport of the active complex into the nucleus to affect downstream pathways. NF-κB can be activated by exposure to lipopolysaccharides, inflammatory cytokines such as tumor necrosis factor-α (TNF-α) or interleukin-1 (IL-1), oxidants, free radicals, inhaled particles and other stimuli (15). Tian et al. showed that AhR can physically associate with RelA (p65 subunit) of NF-κB (16). In vitro and in vivo studies demonstrated that AhR agonists can also activate the NF-κB pathway by inducing nuclear translocation of RelA and p50 (17, 18). Chen et al. found that after activation of AhR by TCDD treatment, Rel A forms a complex with p50, while AhR is associated with ARNT in the nucleus (19). However, after AhR overexpression, AhR is complexed with RelA and not ARNT in the nucleus. Thus, it appears that activated AhR and overexpression of AhR modulate NF-κB activation by different mechanisms. Recent studies also showed that AhR interacts with the non-canonical pathway of NF-κB and its RelB subunit, which plays a key role in the heightened inflammatory response to cigarette smoke. In AhR-knockout mice, there is increased release of pro-inflammatory cytokines IL-6 and TNF-α in bronchoalveolar (BAL) cells, increased expression of cyclooxygenase-2 (COX-2) and prostaglandins (PGs) in lung fibroblasts, and loss of nuclear RelB (20, 21). This finding will be discussed in more detail below.
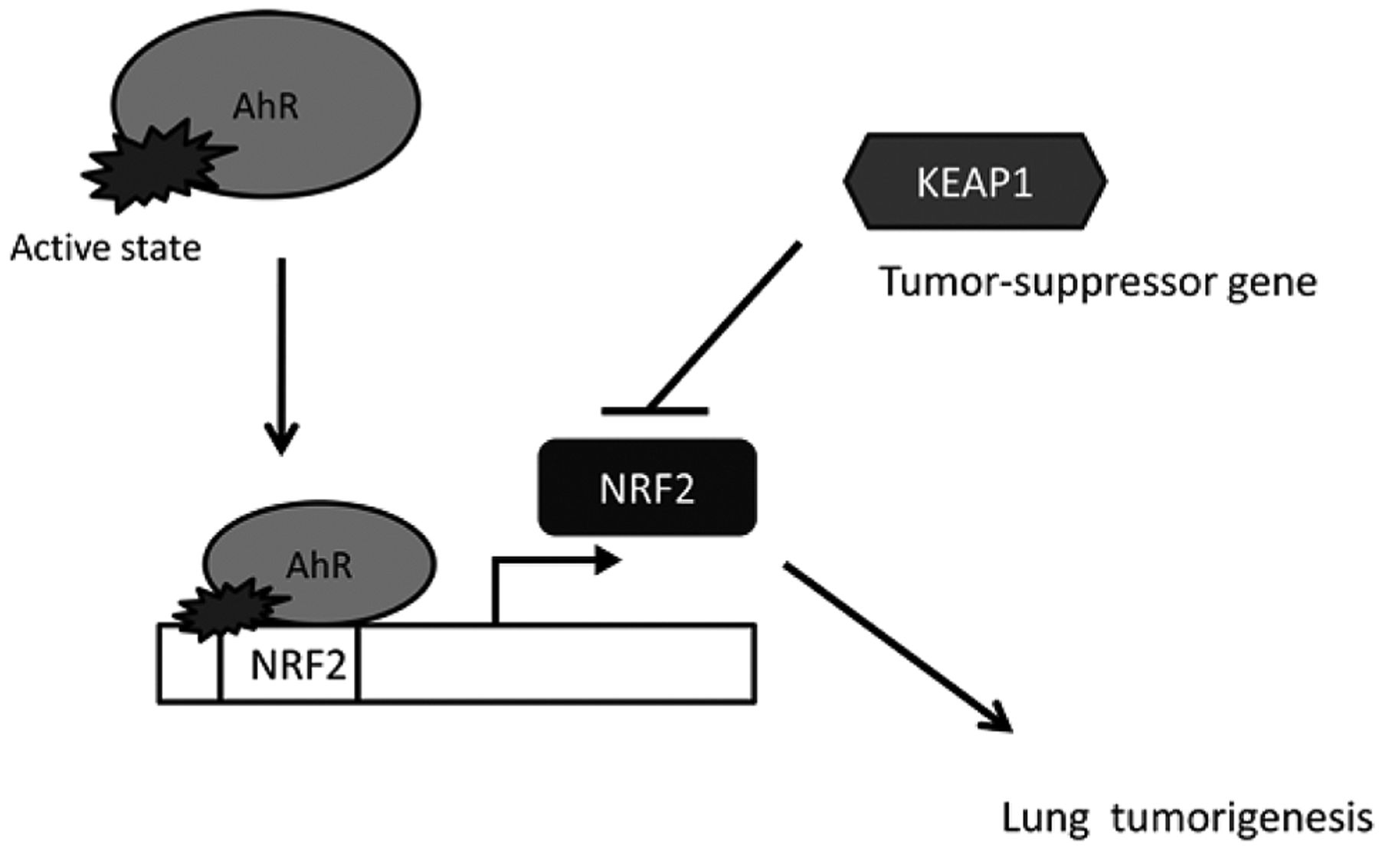
Nuclear factor erythroid 2 p45-related factors (NRF2). A recent area of investigation is the cross-talk between AhR and NRF2. NRF2, a transcription factor which interacts with a diverse array of cytoprotective genes and is involved in the antioxidant response element (ARE) pathway (22, 23). Studies in AhR-deficient cells demonstrated that induction of NRF2 transcription by TCDD is directly regulated by AhR activation (24). In the AhR-deficient Tao cell line (variants of murine hepatoma 1c1c cells), after treatment with 10 nM TCDD for 16 h, quantitative reverse transcription polymerase chain reaction (RT-PCR) showed that NRF2 mRNA expression was nearly abolished when compared to a CYP1A1-deficient cell line (c37). Using a chromatin immunoprecipitation assay, this study also demonstrated that AhR binds directly to the NRF2 promoter region after TCDD exposure (Figure 2). In addition, silencing of AhR expression with siRNA hindered NRF2 mRNA induction by TCDD.
Estrogen receptor (ER). Controversy has surrounded the issue of the incidence of lung cancer in women, as compared to men (25, 26). Female smokers have higher CYP1A1 expression and more DNA adducts in lung tissues compared to male smokers (27, 28). In lung adenocarcinoma cell lines after exposure to cigarette smoke condensate, BaP-DNA adducts and CYP1A1 gene expression were both increased (29) in cell lines of female origin compared to those of male origin. Case-control studies have shown women with the same level of tobacco exposure as men to be at higher risk for lung cancer, with a disproportionately increased risk for adenocarcinoma (28, 30-32). These differences may be due to variabilities in ER activation. When AhR is activated by TCDD or 2-methylcholanthrene (2MC), the AhR/ARNT heterodimer directly associates with ER-α and ER-β, leading to drastic reduction in endogenous ER-α and ER-β (33, 34). Thus, AhR modulation of estrogen signaling may be a key to understanding the difference in lung cancer risk between men and women.
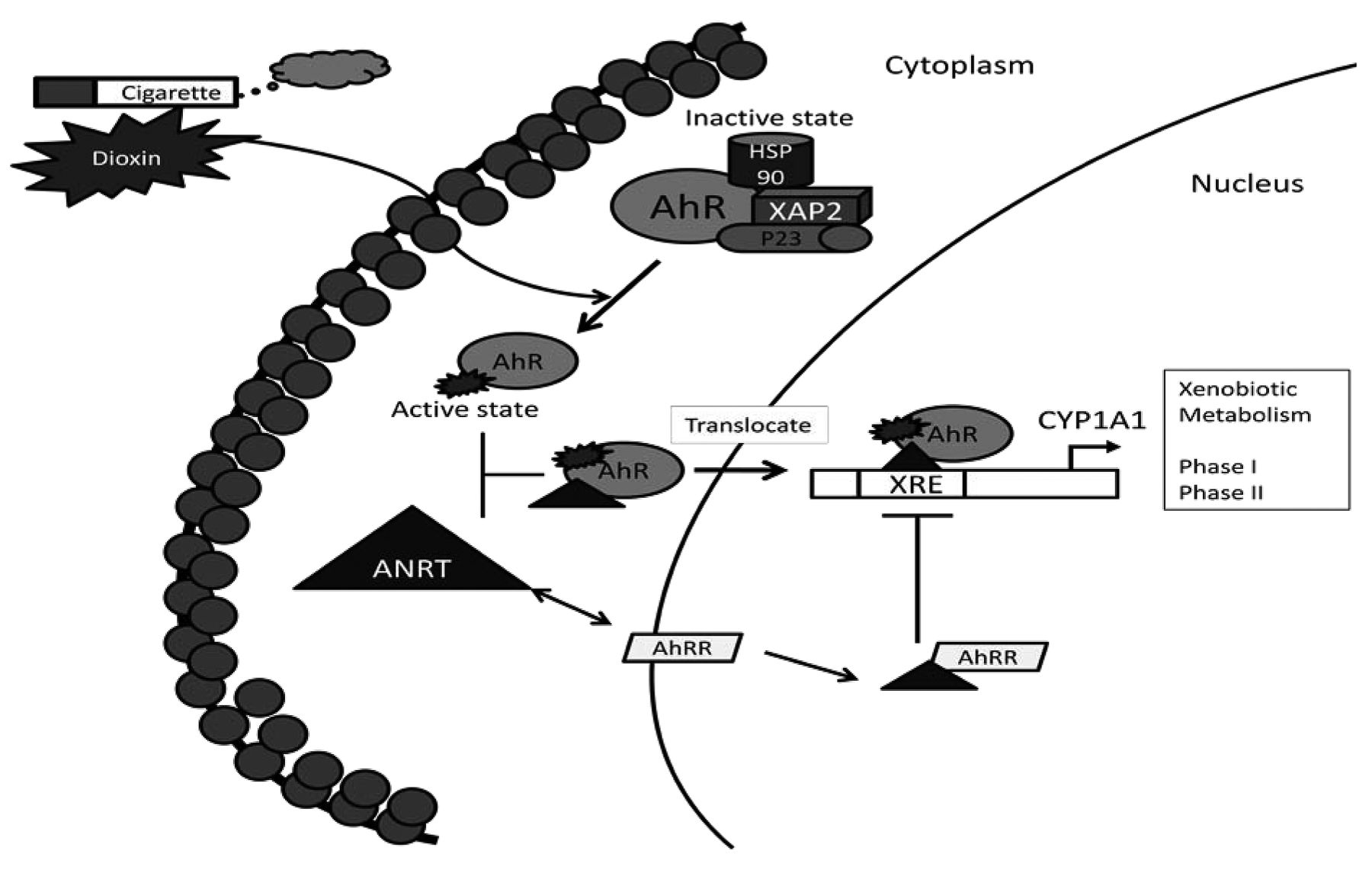
Aryl hydrocarbon receptor (AhR) activation. After binding of AhR ligand, AhR forms a complex with AhR nuclear translocator (ARNT) and translocates into the nucleus where it bind to the xenobiotic response elements (XRE), activating the xenobiotic metabolism. AhR repressor (AHRR) represses AhR transcription by competing with ARNT dimerization. Heat shock protein 90 kDa (HSP90). X-associated protein-2 (XAP2). Prostaglandin E synthase-3 (P23). Cytochrome P450, family-1, member-A1 (CYP1A1).
Osteopontin. Osteopontin is a glycol-phosphoprotein secreted by transformed malignant epithelial cells, and has been studied as a biomarker for the progression and metastasis of lung cancer (35-37). Plasma osteopontin levels are found to be significantly lower after surgical resection of non-small cell lung cancer (NSCLC) (38). Its role in NSCLC pathogenesis is not completely understood, but overexpression of different osteopontin isoforms leads to different degrees of proliferation, colony formation, Matrigel invasion activity, and VEGF secretion compared to controls in NSCLC cell lines (39, 40). Chuang et al. demonstrated that in 72 lung cancer samples, 79% with highly-expressed AhR also expressed osteopontin (41). Similar results were seen in H1355 lung cancer cell lines. Furthermore, using a luciferase reporter assay system, the authors demonstrated that both ligand-independent and ligand-activated AhR activated the osteopontin promoter region and increased osteopontin expression. In particular, inhibition of NF-κB activity, via siRNA knock-down of the p65 subunit of NF-κB, reduced ligand-activated, AhR-mediated induction of the osteopontin promoter.

Aryl hydrocarbon receptor (AhR) and nuclear factor erythroid-2 p45-related factors (NRF2). After exposure to 2,3,7,8-tetrahloro-dibenzodioxin (TCDD), AhR is activated to bind to the NRF2 promoter region, which triggers downstream lung tumorigenesis. Kelch-like (ECH)-associated protein-1 (KEAP1), a tumor-suppressor gene inhibits NRF2 expression.
AhR and Lung Tumorigenesis
The role of AhR in tumorigenesis has been studied in many types of cancers, including breast, liver, ovary, prostate, glioma, and skin (42, 43). For example, in vivo studies of skin tumors in AhR-deficient mice showed that inducibility of CYP1A2 was lost and BaP-induced carcinogenesis was attenuated (44). In mice with ARNT specifically deleted in the epidermis, BaP-induced skin tumors were completely prevented (45). Studies dating back to the 1970s have linked AhR to lung cancer; more recent studies have focused on possible mechanisms of AhR as an initiator of cigarette smoke-induced lung cancer.
Lung tumor initiation through AhR and cytochrome P450. AhR, and specifically its effect on cytochrome P450, is important for the initiation of cigarette smoke-induced lung cancer. Expression levels of different members of the cytochrome P450 family are directly influenced by cigarette smoke. CYP1A1 and CYP1B1 are expressed in human alveolar type I and II cells, ciliated columnar bronchoalveolar epithelial cells, and alveolar macrophages. In human lung tissues, expression of CYP1A1 was higher in smokers (15.5 pmol/mg microsomal proteins) and ex-smokers (19.0 pmol/mg) compared to never-smokers (6.0 pmol/mg) (46). Similar results were also found in cells, in bronchoalveolar lavage and bronchial biopsies in smokers versus non-smokers. CYP1B1 expression also follows a similar pattern (47).
Studies of xenobiotic metabolism in lung cancer reveal that AhR plays a central role in particulate matter and cigarette smoke-induced lung carcinogenesis. In a study using AhR +/+ and AhR −/− mice, airborne particulate extract (APE) was applied to the dorsal skin (48). At the 58th week after application of APE, squamous cell carcinoma occurred in 8/17 AhR +/+ mice and CYP1A1 was induced. In contrast, no tumors were found in AhR −/− mice and expression of CYP1A1 was not induced. In a study using human lung cancer tissues, CYP1A1 mRNA expression was detectable in 7/15 (47%) of tumors from current smokers, 2/24 (8%) of tumors from ex-smokers, and 1/4 (25%) of tumors from non-smokers (49). Additionally, in 78 NSCLC samples, there was a positive correlation between expression of AhR and CYP1A1, predominantly in adenocarcinomas (50). Further examination of adenocarcinomas demonstrated that the levels of AhR mRNA and protein were highly-expressed in lung adenocarcinoma tissues and cell lines compared to normal lung tissues and bronchial epithelial cells (51). In this study, among 107 lung adenocarcinomas examined, 49 had high levels of AhR expression, 52 had high levels of CYP1B1 expression, and 40 expressed CYP1A1. Of interest, in the 57 cases of adenocarcinoma with features of bronchioalveolar carcinoma (BAC), highest expression levels of AhR and CYP1B1 were found in mixed-type specimens with invasive features, followed by BAC without invasive features, and the lowest expression of AhR and CYP1B1 was found in normal adjacent bronchial epithelial cells, suggesting that invasiveness also correlates with AhR expression. Interestingly, AhR RNAi clones, si1414-6 and si1414-7, were found to have reduced anchorage-independent growth and reduced levels of intracellular reactive oxygen species.
Another proposed mechanism for the effect of AhR on lung carcinogenesis is AhR-mediated production of DNA adducts. AhR-dependent enzyme induction increased the frequency of DNA adducts in the lung in AhR-responsive mice (52). One study suggested that AhR activation and CYP1A1 induction is required for BaP-induced DNA adduct formation and increased apoptotic activity in human lung cells (53). Furthermore, treatment with resveratrol, an AhR antagonist, repressed BaP carcinogenic activity by reducing DNA adduct formation and apoptosis in mouse lung tissues (53). Conversely, a study using AhR-knockout mice showed that BaP treatment resulted in increased levels of DNA adducts and BaP metabolites in the lung tissue compared to control mice (54). As a plausible explanation, BaP metabolites may act via an AhR-independent pathway to increase levels of DNA adducts in AhR-knockout mice. Further studies are needed to clarify this relationship.
Lung tumorigenesis: AhR and NF-κB interaction. NF-κB is often highly-expressed in lung cancer and has been linked to lung carcinogenesis (55-57). A recent study explored the role of AhR and NF-κB in lung tumor initiation (19). Immunostaining of 200 NSCLC tissue blocks revealed that nuclear Rel A and IL-6 (cytokine regulated by NF-κB) levels correlated with cytosolic and nuclear AhR levels. In addition, in vitro experiments in BEAS-2B and H1355 cell lines with overexpressed AhR levels also showed significant increase in IL-6 mRNA and protein levels, which were down-modulated by a NF-κB inhibitor, Bay117085. Using an immunofluorescence assay, the investigators also demonstrated that when AhR was overexpressed, there was an increase in RelA nuclear translocation, as well as an increase in AhR/RelA complex. AhR/RelA binds to the κB element to increase NF-κB activity, resulting in the up-regulation of IL-6 expression, which is important in lung tumor initiation (58, 59).
Another proposed mechanism for lung tumorigenesis modulated by AhR and NF-κB involves their role in chronic inflammation, which is known to promote cancer through increased levels of COX-2 and PGs (60). Consistent with the latter, immunoblotting of NSCLC samples showed overexpression of COX-2 and PGs (61). Pre-clinical studies in patients with NSCLC have shown that an increased COX-2 level correlated with worse overall survival and aggressive disease. (62) Selective COX-2 inhibitors were also shown to augment the effects of chemotherapy, and possibly suppress angiogenesis (62, 63). In vitro experiments with human lung fibroblasts showed that AhR seems to play a crucial role in regulating cigarette smoke-related induction of COX-2 and PG pathways (64). These studies demonstrated that TCDD induced COX-2 production in human lung fibroblasts, and an AhR antagonist prevented cigarette smoke extract-induced AhR-ARNT dimerization and nuclear translocation, which in turn reduced the COX-2 level and PG production.
The role of AhR in chronic inflammation has still not been completely elucidated. As noted above, one study using AhR-knockout mice suggested that the physiological role of AhR was intact to counteract inflammation through the NF-κB subunit RelB (20). In AhR-knockout mice, there was increased release of pro-inflammatory cytokines IL-6, TNF-α and PGs in BAL cells when compared to wild-type mice. This study also suggested that the lack of AhR can lead to a rapid loss of RELB in both nuclear fractions of lung cells and BAL cells after exposure to cigarette smoke. In a follow-up study, lung fibroblasts from AhR −/− mice had lower RelB and increased COX-2 and PGs levels compared with control mice after exposure to cigarette smoke extract (CSE) (21). When the expression of AhR was restored in AhR −/− lung fibroblasts through transfection, COX-2 and PG levels were reduced compared to non-transfected AhR −/− fibroblasts. Of note, AhR+/+ lung fibrosis did not exhibit a loss of RelB, but did show a moderately increased expression of COX-2. Hence, additional studies are needed to clarify AhR and NF-κB interaction; however, the evidence seems to suggest an interconnected role in lung tumorigenesis.
AhR and NRF2. Recent advances have been made in understanding the role of NRF2 in lung tumorigenesis (65, 66). One study found an increased incidence of a somatic mutation in the coding region of NRF2 (a type of mutation that results in aberrant cellular accumulation of NrRF2) in patients with squamous cell lung carcinoma compared with smokers without cancer (65) Kelch-like (ECH)-associated protein-1 (KEAP1), a cytosolic repressor of NRF2 located on chromosome 19p13, is considered to be a tumor-suppressor gene. Loss of 19p has been identified in lung adenocarcinomas and asbestos-related lung cancer. For example, a study analyzing copy number aberrations using single-nucleotide polymorphism microarrays identified ethnic-specific areas of genomic changes: in Western European patients with stage I lung adenocarcinoma, there were higher rates of genomic loss from 19p13.3 and 19p13.11; in East-Asian patients there were higher rates of copy number gain on 16p13.13 and 16p13.11 (67). Similarly, using array comparative genomic hybridization and fluorescence in situ hybridization, increased loss of chromosomal region 19p13 was found in tumors of asbestos-exposed patients, and increased frequency of 19p fragment was found in BEAS-2B cells after exposure to crocidolite asbestos for 48 h (68).
In 304 lung tumor tissues, 26% of NSCLC had positive nuclear NRF2 expression. Significantly more squamous cell carcinomas (38%) than adenocarcinomas (18%) had positive expression (69). Low or lack of KEAP1 expression was found in 56% of NSCLCs, including 62% of adenocarcinomas and 46% of squamous cell carcinomas. Analysis also showed that NRF2 expression (hazard ratio=1.75) or low/lack of KEAP1 expression (hazard ratio=2.09) was associated with poor overall survival (69). When mouse lewis lung carcinoma (3LL) cells were implanted in Nrf2-deficient mice, an increased number of pulmonary metastatic nodules was seen compared to wild-type mice (70). This possible anti-metastatic effect of Nrf2 was further explored using KEAP1-knockdown mice with constitutively elevated Nrf2 levels. These mice were resistant to metastasis after 3LL cell implantation. Some studies have shown that mutation or inactivation of KEAP1 is frequently present in NSCLC (71-73). At least one molecular alteration in KEAP1 was found in 57% of the NSCLC cases, with aberrant methylation at the KEAP1 gene promoter being the most common (71). One-third of the NSCLC cases had two alterations, and this was associated with a higher risk of disease progression (hazard ratio=3.62). KEAP1 mutation was seen most frequently in papillary adenocarcinoma (60%), compared to only 18.5% in other NSCLCs. In a separate study, all the KEAP1 mutations were found in tumors from smokers, suggesting a possible interaction with AhR (73). A structural study showed that a point-mutation reduced the affinity of KEAP1 for NRF2 in human lung cancer cells (74). The G430C somatic mutation in KEAP1 was discovered in patients with lung cancer, and the G364C homozygous mutation was found in H1648 lung adenocarcinoma cell line and in the H1148 small cell lung cancer cell line. The substitution of glycine in the G364C mutation affects the structural conformation with the neighboring residue, thereby abolishing the KEAP1-NRF2 interaction. These mutations were also found to reduce cytoplasmic localization of NRF2, and allow constitutive activation. Constitutive NRF2 activation, previously shown to stimulate lung cancer cell replication was achieved through increased peroxiredoxin-1 (PRDX1) expression. PRDX1 is a target gene that enhances cell growth and radio resistance of A549 cells (66). Greater understanding of the interaction between AhR and Nrf2 will shed more light on their roles in lung tumor development.
AhR and Tumor Growth and Metastasis
Hanahan and Weinberg proposed six hallmarks of cancer to help understand and organize the complexity of the biology of tumors (75). These hallmarks consist of sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis. Recent studies suggest that AhR likely plays an important role in many of these hallmarks of cancer.
AhR, cell cycle, and evasion of apoptosis. AhR may play a role in carcinogenesis through its effect on cell proliferation. For example, in A549 cells (human adenocarcinoma cell line), both a direct AhR agonist and overexpression of AhR-stimulated cell proliferation by affecting the cell cycle (76). β-Naphthoflavone, an AhR ligand, stimulated A549 growth; on the contrary, α-naphthoflavone (also an agonist, but unable to transform AhR to its DNA-bound form) had no effect on A549 growth. These findings suggest that DNA binding to AhR is needed for cell proliferation. Using flow cytometry to analyze the role of AhR in cell-cycle progression, the investigators found that 10% of AhR-overexpressing cells were in the S phase and none were in G2/M phases, while in control cells, 10% were in G2/M and none were in the S phase. In addition, using a cell-cycle array to study 111 genes, the investigators found a 4- to 5-fold increase in the level of transcription factor DP2, replication factor C38 (RFC38), and proliferation cell nuclear antigen (PCNA); replication factor in AhR-overexpressing cells. AhR can also promote tumor proliferation through its interaction with various oncogenes. Ramakrishna et al. found that the levels of the K-Ras-p21 protein in the membranes in mouse lung were highest in the highly AhR-responsive mouse strains. Exposure of AhR-responsive mice to TCDD induced a 3-fold increase in the membrane fraction of K-Ras-p21 in the lung, while AhR non-responsive mice had no change in the K-Ras-p21 levels (77). The AhR-responsive mouse strains have been previously shown to be more susceptible to developing malignancies (78).
AhR and tissue invasion and metastasis. Evidence suggests that AhR mediates de-regulation of cell-cell contact by stimulating migration and epithelial mesenchymal transformation after exposure to TCDD (43). One study demonstrated that dioxin after binding to AhR triggered remodeling of the cytoskeleton of epithelial cells by reducing cell-cell adhesion through a c-Jun NH(2)-terminal protein Kinases (JNK)-dependent pathway (79). Cigarette smoke can affect lung fibroblasts, the cells that produce collagen and the extracellular matrix. Studies of primary lung fibroblasts from AhR −/− mice compared to AhR +/+ fibroblasts showed that after exposure to CSE, AhR −/− cells had lower viability and proliferation, increased caspase activation, and increased cleaved Poly ADP ribose polymerase (PARP) and lamin A/C (markers of apoptotic protein cleavage) (80). Hence, AhR seems to play a crucial role in reducing cell-cell adhesion, which results in an increase in the motility and invasiveness of cancer cells.
AhR and angiogenesis. AhR also plays a role in angiogenesis, the formation of new blood vessels to supply nutrients and oxygen to support the proliferation of tumor cells. Angiogenesis plays a key role in the progression and growth of most types of solid tumors. The proliferation of vascular endothelial cells is controlled by a number of growth factors, transcription factors, and cytokines, including basic fibroblast-like growth factor, transforming growth factor-beta, vascular endothelial growth factor (VEGF), IL-8, angiopoietin-2, and hypoxia-inducible factor-1 (HIF-1). ARNT, also known as HIF-1β, can form a heterodimer with HIF-1α in response to oxygen deprivation (Figure 3). Using mice lacking the ARNT protein, Maltepe et al. showed that during hypoxia, ARNT transcriptional activity is required for VEGF activity, tissue factor production and blood vessel development (81). Using AhR-null mice, they demonstrated that angiogenesis was modulated by AhR through VEGF activation in the endothelium and TGF-β inactivation in the stroma (82).
AhR Polymorphism in Cancer Risk
The association of AhR gene polymorphisms with risk of lung cancer among cigarette smokers is controversial. Studies in Japan, Finland, and France did not show relationships among AhR gene polymorphism, ARNT polymorphism, smoking, CYP1A1 inducibility, and lung cancer (83-87). However, more recently, studies showed that smokers with GGG haplotype single-nucleotide polymorphic sites in the AhR gene had an increased risk (adjusted OR=3.2) for lung cancer, particularly squamous cell carcinoma (88). Two missense mutations (Val323Leu and Arg101Stop) in the human CYP2A13 gene were associated with an elevated risk for small cell lung cancer (OR=9.9) (89). In a case control study in China of 500 patients with lung cancer and 517 cancer-free controls, single-nucleotide polymorphisms rs2158041 and rs7811989 of AhR were associated with lung cancer in heavy smokers (adjusted ORs 1.53 and 1.48, respectively) (90).

Aryl hydrocarbon receptor translocator (ARNT) and angiogenesis. In a hypoxic state, ARNT forms a heterodimer with hypoxia-inducible factor (HIF-1α), activating vascular endothelial growth factor (VEGF), and promoting angiogenesis.
Conclusion
The cytosolic aryl hydrocarbon receptor and its family of proteins closely interact with pro-carcinogens PAHs in cigarette smoke and play an important role in lung carcinogenesis. AhR plays a central role in the metabolism of PAHs through phase-I cytochrome P450 enzymes, and regulates the expression of numerous genes that contribute to the initiation, promotion, and progression of lung cancer. AhR not only mediates its effects via physical association with NF-κB subunits, but also affects downstream signaling pathways that control the initiation and promotion of lung tumorigenesis. New evidence suggests cross-talk with additional transcription factors, including NRF2, and ER. With a better understanding of AhR and its role in lung carcinogenesis, new biomarkers for the early detection of lung cancer may be identified and novel therapies and drugs can be developed to effectively target the AhR pathway to prevent and treat lung cancer.
Acknowledgements
W.N.R. is supported by U01CA086137 and HL090316. J.T. is supported by T32ES007267, UL1 TR000038 and is a recipient of the Stony-Wold Herbert Fund Fellowship.
- Received February 19, 2013.
- Revision received March 19, 2013.
- Accepted March 20, 2013.
- Copyright© 2013 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Degradation of SARS-CoV-2 receptor ACE2 by tobacco carcinogen-induced Skp2 in lung epithelial cells
- Biomarkers of Exposure and Effect in the Lungs of Smokers, Nonsmokers, and Electronic Cigarette Users
- MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer
- The aryl hydrocarbon receptor regulates nucleolar activity and protein synthesis in MYC-expressing cells
- Activation of the Aryl Hydrocarbon Receptor Leads to Resistance to EGFR TKIs in Non-Small Cell Lung Cancer by Activating Src-mediated Bypass Signaling
- PHLPP2 Downregulation Contributes to Lung Carcinogenesis Following B[a]P/B[a]PDE Exposure