


Abstract
Wilms' tumor gene WT1 is highly expressed in leukemia and in various types of solid tumors and exerts an oncogenic function. Thus, WT1 protein is a most promising tumor-associated antigen. We have been successfully performing WT1 vaccination with a 9-mer modified WT1235 peptide, which has one amino acid substitution (M→Y) at position 2 of 9-mer natural WT1235 peptide (235-243 a.a.), for close to 700 HLA-A*24:02-positive patients with leukemia or solid tumors. Although vaccination of modified WT1235 peptide induced natural WT1235 peptide-recognizing cytotoxic T-lymphocytes (CTLs) and exerted cytotoxic activity towards leukemia and solid tumor cells that expressed the natural WT1235 peptide (epitope) but not the vaccinated modified WT1235 peptide (epitope), the molecular basis has remained unclear. In this study, we established a modified WT1235 peptide-specific CTL clone, we isolated T-cell receptor (TCR) genes from it and transduced the TCR genes into CD8+ T-cells. The TCR-transduced CD8+ T-cells produced interferon-γ (IFNγ) and tumor necrosis factor-α (TNFα) in response to stimulation not only with the modified WT1235 peptide but also with the natural WT1235 peptide and lysed modified or natural WT1235 peptide-pulsed target cells and endogenously WT1-expressing leukemia cells in a HLA-A*24:02-restriction manner. These results provided us, for the first time at molecular basis, with a proof-of-concept of modified WT1235 peptide-based immunotherapy for natural WT1235 peptide-expressing malignancies.
It is evident that T-cell-mediated immunity plays a crucial role in tumor regression and eradication, and the main effector cells that attack tumor cells are CD8+ cytotoxic T-lymphocytes (CTLs) (1, 2). These CTLs recognize tumor-associated antigen (TAA)-derived peptides presented on the surface of target cells in association with major histocompatibility complex (MHC) class I molecules. To enhance the activity of the TAA-specific CTLs, various types of immunotherapies, including cancer vaccines, are being performed (3, 4).
WT1, which was originally identified as a gene responsible for the pediatric neoplasm Wilms' tumor, encodes a zinc finger transcription factor involved in the regulation of cell proliferation and differentiation (5-8). Although the WT1 gene was first categorized as a tumor suppressor gene, we showed that it had an oncogenic function and the WT1 protein was highly expressed in various kinds of malignant neoplasms, including hematopoietic malignancies and solid tumors, indicating that the WT1 protein is a most promising TAA (9-21).
Our group and others have identified WT1 protein-derived CTL epitope peptides with the restriction of several HLA class I types. Clinical trials using WT1 CTL epitopes, including HLA-A*0201-restricted WT1126 and HLA-A*24:02-restricted WT1235 peptides, were performed and showed successful results with clinical response (22, 23). However, we identified a modified WT1235 peptide with much higher affinity for HLA-A*24:02 than the natural WT1235 peptide. The modified WT1235 peptide was found to have the ability to elicit robust induction of the peptide-specific CTLs that also recognized the natural WT1235 peptide (epitope) presented on the tumor cell surface (24). In fact, vaccination of the modified WT1235 peptide, which was mainly conducted by our group, showed favorable clinical response, including tumor shrinkage and leukemia cell reduction, in association with immunological response, such as an increase in the frequency of natural WT1235 peptide-specific CD8+ T-cells in the peripheral blood (PB) of patients with various kinds of malignancies (3, 25-36). However, why the vaccination of modified WT1235 peptide exerted clinical effect and killed tumor cells that expressed the natural WT1235 peptide (epitope) but not the modified WT1235 peptide (epitope) has not yet been explained on a molecular basis.
In the present study, we describe the establishment of a modified WT1235 peptide-specific CTL clone, the isolation of the T-cell receptor (TCR) genes from it, and the molecular basis of clinical findings that the vaccination of modified WT1235 peptides is effective for eradication of natural WT1235 peptide (epitope)-expressing tumor cells.
Materials and Methods
Cells. Peripheral blood mononuclear cells (PBMCs) were obtained from a healthy donor with HLA-A*24:02 by density gradient using a lymphocyte separation solution (Nacalai Tesque, Kyoto, Japan), and CD8+ T-cells were isolated from the PBMCs using the Human CD8 T-Lymphocyte Enrichment Set-DM (BD Biosicences, San Jose, CA, USA).
K562 is a cell line derived from a blast crisis of chronic myeloid leukemia (CML). K562 endogenously expresses WT1, but does not express HLA molecules on the cell surface. K562/24:02 is an HLA-A*24:02-expressing K562 cell line, which was generated by the transduction of HLA-A*24:02 cDNA into K562 (37). T2 is a cell line deficient in transporter-associated with antigen processing (TAP) protein that is essential for the transportation and presentation of peptides generated from endogenous proteins. T2/24:02 was made by the transduction of HLA-A*24:02 cDNA into T2 cells (38). K562, K562/A24:02, and T2/A24:02 cells were cultured in RPMI-1640 (Nacalai Tesque), supplemented with 10% fetal bovine serum (FBS; EuroClone, Pero, Italy).
Induction of the modified WT1235 peptide-specific CD8+ T-cell clones. Modified WT1235 peptide (CYTWNQMNL)-specific CD8+ T-cell clones were generated by a mixed lymphocyte peptide culture (MLPC) in a modification of the method described by Karanikas et al. (39). PBMCs from an HLA-A*24:02+ healthy donor were cultured in X-VIVO 15 medium (Lonza, Walkersville, MD, USA), supplemented with 10% human AB type serum (GEMINI Bio-Products, West Sacramento, CA, USA) in the presence of the modified WT1235 peptide (1 μg/ml) and recombinant interleukin-2 (IL-2) (40 U/ml, kindly donated by Shionogi & Co., Ltd., Osaka, Japan) in a 96-well U-bottom plate at a density of 2×105 cells/well so that cell expansion occurred in fewer than 10 wells among 96 wells (39).
After two weeks of culture, the expanded cloned cells were screened for positivity for the phycoerythrin (PE)-conjugated modified WT1235 peptide tetramer (MBL, Nagoya, Japan) and positive clones were confirmed for the peptide specificity by peptide-specific interferon-γ (IFNγ) production.
Cloning of TCR cDNA and construction of a lentivirus vector. cDNA was obtained by reverse-transcription of total mRNA of the modified WT1235 peptide-specific CD8+ T-cell clone B10. cDNAs of TCR-α and -β chains were cloned, amplified by 5’RACE PCR using SMARTer™ RACE cDNA Amplification Kit (Clontech Laboratories, Inc., Mountain View, CA, USA) with gene-specific primers of TRAC (CTGTCTTACAATCTTGCAGATC) for TCR-α chain, and TRBC1 (CACTTCCAGGGCTGCCTTC) and TRBC2 (TGACCTGGGATGGTTTTGGAGCTA) for TCR-β chain, and sequenced.
To construct a vector that simultaneously expressed both the TCR-α and -β chains, cDNAs of the TCR-α and -β chains were linked via a viral P2A sequence (40), followed by cloning into a lentiviral SIN vector (CSII-EF-MCS-IRES2-Venus), with the Venus gene that expressed yellow fluorescent proteins (YFPs) (41).
Transduction of TCR construct into CD8+ T-cells. HEK293T packaging cells were transfected with the TCR construct vector, pCAG-HIVgp and pCMV-VSV-G-RSV-Rev using linear polyethyleneimines (Polysciences, Inc., Warrington, UK) in low-serum media (Gibco, Grand Island, NY, USA). The original CSII-EF-MCS-IRES2-Venus mock vector (mock vector) was used as a negative control. After 12 h of incubation, the HEK293T cells were cultured for virus production in DMEM, containing 4.5 g/l glucose (Nacalai Tesque) supplemented with 10% FBS for 48 h. The virus particles were concentrated by precipitating the culture supernatant using polyethylene glycol (SBI, Mountain View, CA, USA).
The TCR genes were transduced into CD8+ T-cells. In brief, CD8+ T-cells were isolated from PBMCs of an HLA-A*24:02+ healthy donor and activated in X-VIVO 15 medium containing a monoclonal antibody (mAb) against CD28 (eBiosicence Inc., San Diego, CA, USA) and 10% human AB type serum in a CD3 mAb (eBiosicence Inc.)-coated culture plate. After 3 days of activation, the cells were infected with the TCR-containing lentivirus vector using 8 μg/ml of polybrene in RetroNectin (TaKaRa, Tokyo, Japan)-coated plate for 12 h, washed, and cultured in X-VIVO 15 medium, supplemented with 10% human AB type serum.
Flow cytometric analysis. For multicolor staining of cells with tetramer and mAbs, the cells were suspended in phosphate-buffered saline (PBS) containing 2% FBS, followed by staining with the PE-conjugated natural or modified WT235 tetramer according to the manufacturer's protocol. The cells were then stained with mAbs on ice for 20 min, washed twice with PBS, containing 2% of FBS, and analyzed with a FACSAria instrument (BD Biosciences). mAbs used were Pacific Blue-conjugated anti-CD3 (BD BioScience), allophycocyanin (APC)-conjugated anti-CD8 (BD BioScience), and PE-conjugated anti-Vβ1 (TRBV9 in another family nomenclature) mAbs (Beckman Coulter Inc., Bera, CA, USA).
Cytokine production assay. For cytokine production assay, 2.5×104 of responder cells were stimulated by the appropriate stimulator cells pulsed with 10 μg/ml of a natural WT1235 peptide (CMTWNQMNL), the modified WT1235 peptide (CYTWNQMNL), or an irrelevant CMV pp65 peptide (QYDPVAALF) in culture medium containing anti-CD28/49d (BD Bioscience) and 10 μg/ml Brefeldin A for 5 h. After the stimulation, the responder cells were stained with APC-Cy7-conjugated anti-CD8 mAb, washed twice, fixed, and permeabilized with Cytofix/Cytoperm (BD Bioscience). The cells were then stained by a PE-conjugated anti-IFNγ and APC-conjugated anti-TNFa mAbs (BD Bioscience), and analyzed using a FACSAria instrument.

Establishment of a modified WT1235 peptide-specific CD8+ T-cell clone, B10. B10 cells were stimulated by the modified WT1235 peptide, the natural WT1235 peptide, or not stimulated. Flow cytometory of interferon-γ (IFNγ) production by B10 cells is shown.
For HLA blocking assay, an appropriately titrated blocking mAb for HLA class I (clone wb/32) or HLA-DR (clone L243) was added to cell culture for cytokine production assay.
Cytotoxicity assay. Target cells for cytotoxicity assay were labeled with 51Cr in X-VIVO 15 medium, supplemented with 1% human AB type serum for 2 h, and washed with PBS. The target cells were incubated with appropriate concentrations of antigen peptides, if needed. TCR-transfected CD8+ T-cells were co-cultured with the 51Cr-labeled target cells in X-VIVO 15 medium supplemented with 1% human AB type serum for 4 h. The supernatant was collected, and the radioactivity was counted using a MicroBeta2 plate counter. The percentage-specific lysis was calculated by the equation: (cpm experimental release – cpm spontaneous release)/(cpm maximum release – cpm spontaneous release).
Results
Establishment of a modified WT1235 peptide-specific CD8+ T-cell clone. PBMCs of an HLA-A*24:02+ healthy donor were stimulated with modified WT1235 peptide seeded at concentrations of 2×105 cells/well in a 96-well plate and then cultured in the presence of modified WT1235 peptide (1 μg/ml) and IL-2 (40 IU/ml) for two weeks. Cell expansion was observed in only two of a total of 192 wells and finally only one clone, designated B10, was established. B10 cloned cells were positive for staining with HLA-A*24:02/modified WT1235 tetramer and produced IFNγ on stimulation with not only modified WT1235 but also natural WT1235 peptides (Figure 1). These results show that B10 was a modified WT1235 and natural WT1235 peptide-specific CD8+ T-cell clone.
Isolation of the TCRs from B10 and establishment of the TCR-transfected CD8+ T-cells. cDNA of TCR-α and -β chains was made from mRNA of the B10 cells using each gene-specific primer, cloned, and sequenced. V- and J- regions of Vα were TRAV27*01 and TRAJ28*01, respectively, while V-, D-, and J-regions of Vβ were TRBV9*01, TRBD2*01, and TRBJ2-3*01, respectively. The TCRs isolated from B10 cells are referred to as B10-TCRs in the following text.
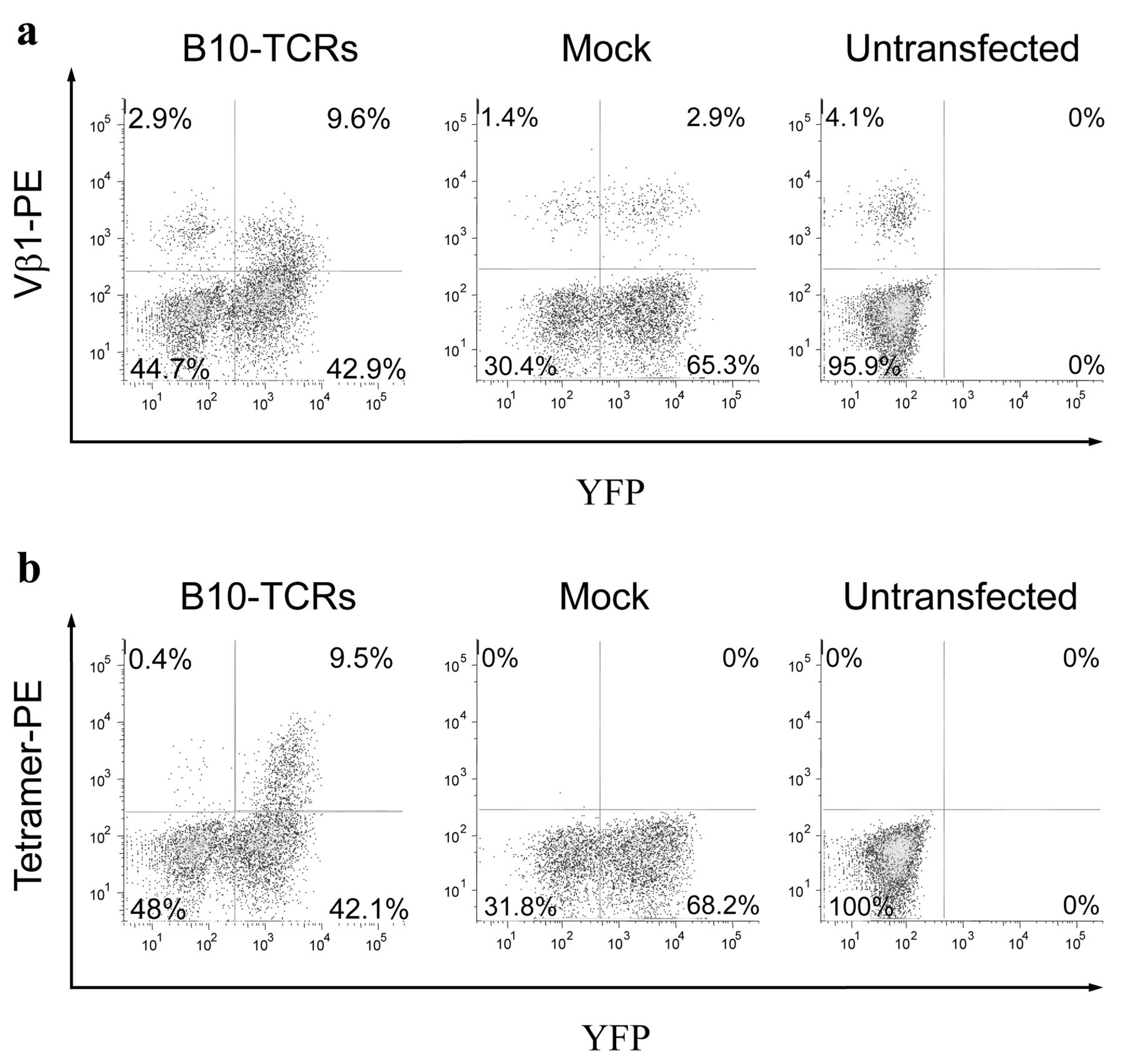
Next, the TCR-α and -β chain genes were linked via a viral P2A sequence for dual gene expression (40) and inserted into a lentiviral vector for transfection. Activated CD8+ T-cells were transfected with a B10-TCR-containing lentiviral vector, stimulated by irradiated autologous PBMCs loaded with modified the WT1235 peptide three days after transfection, cultured for two weeks, and stained with mAbs to CD3, CD8 and either of the anti-Vβ1 family mAb and the modified WT1235-tetramer (Figure 2).
A considerable proportion (18.3%, 9.6/(9.6+42.9)) of YFP-positive cells in B-10-TCR-transfected CD8+ T-cells were positive for staining with mAb to Vβ1 (=TRBV9), whereas 4.3% (2.9/(2.9+65.3)) of YFP-positive cells in mock-transfected CD8+ T-cells were positive for staining with the mAb against Vβ1 mAb (Figure 2a). On the other hand, 4.1% (4.1/(4.1+95.9)) of the untransfected CD8+ T-cells were stained with mAb to Vβ1, which suggested endogenous expression of Vβ1 and/or artificial staining with mAb to Vβ1. Furthermore, importantly, modified WT1235 tetramer-positive cells were detected only in B10-TCR-transfected CD8+ T-cells at frequencies of 18.4% (9.5/(9.5+42.1)) in YFP-positive cells (Figure 2b). These results indicate that the TCRs from B10 were successfully transduced into CD8+ T-cells and were functional.

Functional expression of B10-TCR genes in CD8+ T-cells. Activated CD8+ T-cells were transfected with B10- TCR-containing a lentivirus vector or a mock vector, and then stained with a monoclonal antibody to Vβ1 family (a) or modified WT1235-tetramer (b). Representative data of three experiments are shown.
To assess the function of B10-TCRs, the antigen-specific cytokine production from the CD8+ T-cells transfected with B10-TCRs was examined (Figure 3a and b). B10-TCR-transfected CD8+ T-cells were stimulated by irradiated autologous PBMCs loaded with the modified WT1235 peptide for two weeks and then stimulated again with modified, natural WT1235 peptide, or irrelevant CMV pp65 peptide for 5 h and examined for production of IFNγ and TNFα. Cells stimulated with the modified or natural WT1235 peptide produced IFNγ and TNFα, whereas cells stimulated with the irrelevant peptide (CMV pp65) did not.
Next, HLA class I restriction of B10-TCR-transfected CD8+ T-cells was examined (Figure 3c). The B10-TCR-transfected CD8+ T-cells were stimulated by T2/24:02 cells loaded with the modified WT1235 peptide in the presence of an HLA class I or HLA DR blocking mAb and stained for intracellular IFNγ. The production of IFNγ was inhibited by anti-HLA class I mAb, but not by anti-HLA DR blocking mAb. These results indicate that the cytokine production of B10-TCR-transfected CD8+ T-cells by antigenic stimulation was restricted to HLA class I.

Cytokine production by the stimulation of B10-TCR-transfected CD8+ T-cells. a: B10-TCR-transfected CD8+ T-cells were stimulated with the indicated antigen peptides and examined for IFNγ and TNFα production. Representative data of two experiments is shown. b: Frequencies of intracellular IFNγ- and TNFα-positive cells among YFP-positive cells in B10-TCR-transfected CD8+ T-cells, stimulated with the indicated antigen peptides. c: CD8+ T-cells transfected with the B10-TCRs were stimulated with the modified WT1235 peptide-loaded T2/24:02 cells, and were assayed for IFNγ production in the presence of HLA class I- or HLA DR-blocking monoclonal antibody. Representative data of two experiments are shown. T2/24:02 cells, HLA-A*24:02-positive T2 cells.
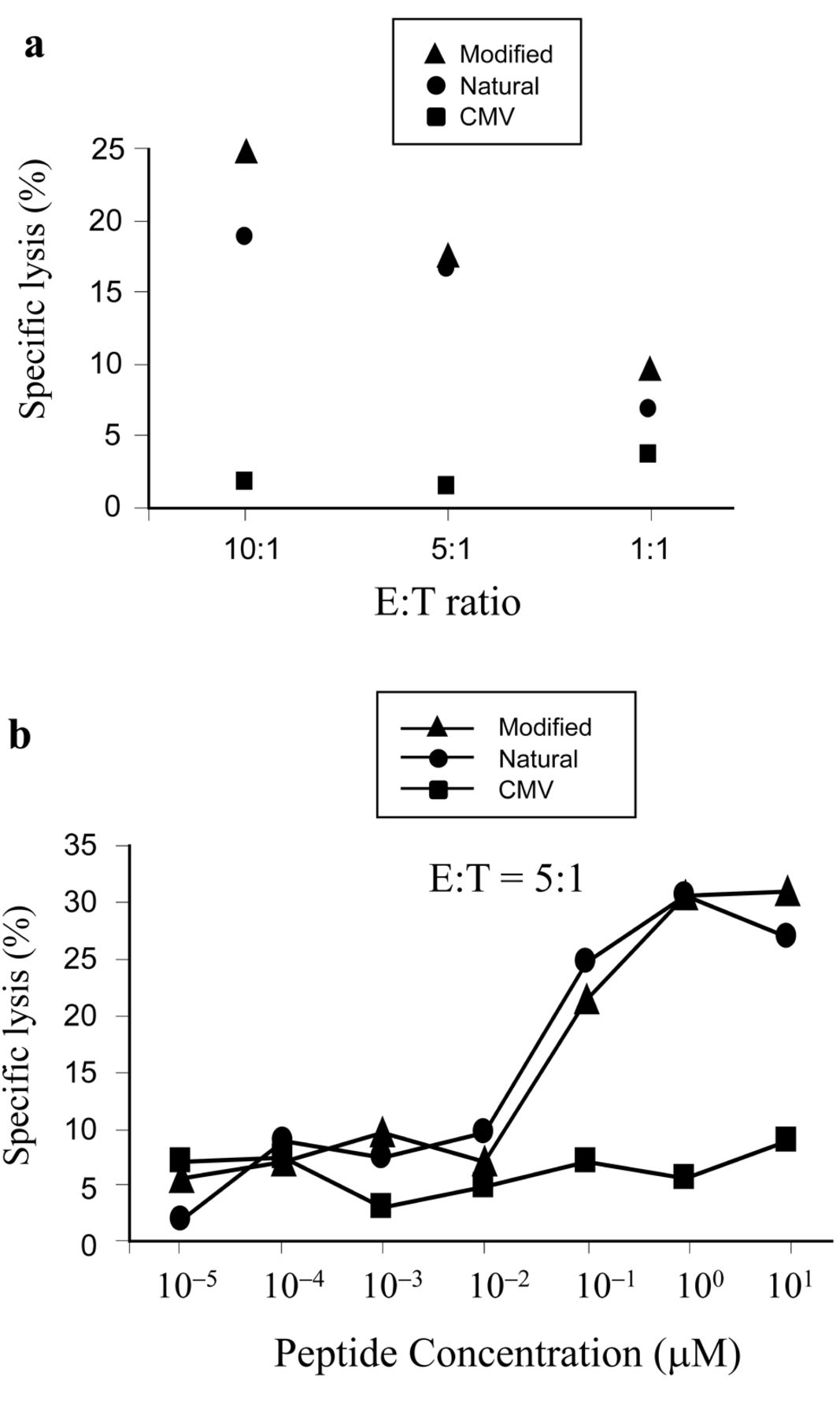
WT1235 peptide-specific cytotoxic activity of B10-TCR-transfected CD8+ T-cells. CD8+ T-cells transfected with B10-TCRs were assayed for their cytotoxic activity towards T2/24:02 cells loaded with modified, natural WT1235 peptide, or with CMV pp65 peptide, at a concentration of 20 μM (a), or at different concentrations (b). Representative data of two experiments are shown. E:T, effector/target ratio.
WT1235 peptide-specific cytotoxic activity of B10-TCR-transfected CD8+ T-cells. To test the antigen-specific cytotoxicity of B10-TCR-transfected CD8+ T-cells, they were co-cultured with irradiated autologous PBMCs loaded with modified WT1235 peptide for two weeks and assayed for cytotoxicity towards 51Cr-treated HLA-A*24:02-transfected T2 (T2/24:02) cells, loaded with modified or natural WT1235 peptide (Figure 4a). The B10-TCR-transfected CD8+ T-cells exhibited cytotoxicity towards the T2/24:02 cells loaded with modified or natural WT1235 peptide in an effector/target (E/T) ratio-dependent manner, but not towards those loaded with an irrelevant peptide (CMV pp65 peptide). These results indicated that B10-TCRs recognized not only the modified WT1235 peptide/HLA-A*24:02 complex but also the natural WT1235 peptide/HLA-A*24:02 complex.

Cytotoxic activity of B10-TCR-transfected CD8+ T-cells towards endogenously WT1-expressing leukemia cells. Cytotoxic activity of B10-TCR-transduced CD8+ T-cells towards endogenously WT1-expressing K562 leukemia cells with or without HLA-A*24:02 expression was examined. Representative data of three experiments are shown.
Next, specific lysis by B10-TCR-transfected CD8+ T-cells was assayed for the T2/24:02 target cells pulsed with different concentrations of modified or natural WT1235 peptide (Figure 4b). The specific lysis increased in parallel with an increase in the peptide concentrations and reached a plateau at an E/T ratio of 5:1, at a concentration of 1 μM in both peptides. The half-maximal lysis for modified and natural WT1235 peptide was obtained at a concentration of about 0.06 μM and 0.04 μM, respectively. These results indicate that the affinity of B10-TCRs for natural WT1235 peptide/HLA-A*24:02 complex was high enough to expect that B10-TCRs would be able to recognize the endogenous WT1 protein-derived (natural) WT1235 peptide that was presented on the cell surface in association with HLA-A*24:02 molecules.
Lysis of endogenously WT1-expressing leukemia cells by B10-TCR-transfected CD8+ T-cells with an HLA-A*24:02 restriction. Whether or not B10-TCR-transfected CD8+ T-cells had the ability to lyse endogenously WT1-expressing leukemia cells with a restriction of HLA-A*24:02 was examined. The B10-TCR-transfected CD8+ T-cells were stimulated by irradiated autologous PBMCs loaded with the modified WT1235 peptide. After two weeks of the stimulation, the B10-TCR-transfected CD8+ T-cells were assayed for the lysis of HLA-A*24:02-transfected K562 leukemia cells (K562/24:02) that endogenously expressed WT1. The B10-TCR-transfected CD8+ T-cells were cytotoxic towards the K562/24:02 cells, but not towards K562 cells without an HLA-A*24:02 expression (Figure 5). These results indicate that B10-TCR-transfected CD8+ T-cells were able to kill endogenously WT1-expressing leukemia cells in an HLA-A*24:02 restriction manner.
Discussion
In the present study, a modified WT1235 peptide-specific CTL clone (B10) was established and its TCRs (B10-TCRs) were cloned. B10-TCR-transfected CD8+ T-cells were able to kill both modified WT1235 peptide-pulsed and natural WT1235 peptide-pulsed target cells and endogenously WT1-expressing leukemia cells.
An important finding presented here was that B10-TCRs, isolated from a modified WT1235 peptide-specific CTL clone, was able to recognize and kill both natural WT1235 peptide-pulsed target cells and endogenously WT1-expressing leukemia cells that were possibly expressing natural WT1235 peptide (epitope) on their cell surface in complexes with HLA-A*24:02 molecules. The evidence, at the molecular level, showing that a modified WT1235 peptide-specific TCR recognizes both its own modified and other natural WT1235 peptides (epitopes) has been demonstrated here for the first time due to our successful cloning a modified WT1235 peptide-specific TCR gene. This evidence provided us with a strong proof-of-concept of modified WT1235 peptide-based immunotherapy, in which the modified (not natural) WT1235 peptides were effectively vaccinated for the eradication of tumor cells that were possibly expressing natural (not modified) WT1235 peptides in complexes with HLA-A*24:02 molecules. In fact, there are some clinical findings showing that vaccination with modified WT1235 peptides induced modified WT1235 peptide-specific CTLs and other CTLs that were able to recognize both the modified and natural WT1235 peptides (epitopes). For example, Narita et al. successfully vaccinated a patient with CML with the modified WT1235 peptides and showed that some CD8+ T-cells in PBMCs that were obtained after repeated WT1 vaccination were dually stained with the modified WT1235 peptide-specific and natural WT1235 peptide-specific tetramers. They also showed that the modified WT1235 peptide-specific CTL clones established, exerted cytotoxic activity towards both the modified WT1235 peptide-pulsed and natural WT1235 peptide-pulsed target cells (42). However, since the cloning of TCRs from the modified WT1235-specific CTLs was not done, it was not demonstrated, at the molecular level, that the TCRs of the modified WT1235-specific CTLs recognized both the modified and natural WT1235 peptides (epitopes). On the other hand, it was demonstrated that a natural WT1235 peptide-specific CTL clone, TAK-1, recognized both the natural and modified WT1235 peptides (24). However, the molecular basis of this finding has not yet been reported. Thus, detailed analysis at the molecular level for explaining how WT1235 peptide-specific CTLs are able to recognize both natural and modified WT1235 peptides (epitopes) has been reported here for the first time.
Results presented here suggest the possibility for adoptive transfer therapy of CD8+ T-cells transfected with the modified WT1235 peptide-specific TCR genes. Half-maximal lysis by the CD8+ T-cells that were transfected with the TCRs from the modified WT1235 peptide-specific CTLs was obtained against the natural WT1235 peptide-pulsed target cells at concentrations of as low as 0.04 μM. This indicates the high affinity of the TCRs for the natural WT1235 epitope on tumor cells. These results should allow us to expect a good clinical effect of adoptive cell therapy using the TCR genes isolated here.
Acknowledgements
This study was supported in part by a Grant-in-Aid from the Ministry of Education, Science, Sports, Culture, and Technology, Japan.
- Received September 20, 2012.
- Revision received October 22, 2012.
- Accepted October 23, 2012.
- Copyright© 2012 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}