


Abstract
Background: Acute myeloid leukemia (AML) cases with t(8;21) or inv(16) have a favorable outcome, but the associated prognoses are heterogeneous and complicated by additional molecular aberrations. Patients and Methods: Between January 2000 to December 2010, 67 patients were diagnosed with t(8;21) or inv(16) AML. We collected cytogenetic variables and analyzed treatment outcomes. Results: Among 67 patients, 51 (7.8%) had t(8;21) AML and 16 (2.4%) had inv(16) AML. Thrombocytopenia, and a high percentage of blasts in the peripheral blood and bone marrow were associated with poor overall survival. Twenty-five (49.0%) patients with t(8;21) had an additional chromosomal abnormality, while only six (37.5%) patients with inv(16) AML had a secondary chromosomal abnormality. The most common chromosomal abnormalities were deletion of the Y or X sex chromosomes. Conclusion: Deletion of the Y chromosome may be a favorable prognostic factor in patients with core binding factor-positive AML.
Core-binding factor (CBF) is a heterodimeric transcription factor complex composed of alpha and beta subunits that plays a pivotal role in normal hematopoiesis (1). CBF-positive acute myeloid leukemia (AML) results from the disruption of the α and β subunits of CBF genes, and is characterized by the presence of t(8;21)(q22;q22) or inv(16)(p13q22)/t(16;16)(p13;q22) (2). The CBFα subunit is encoded by one of the three homologous genes belonging to the Runt-related transcription factor (RUNX) family, the RUNX1 (AML1; CBFA2; PEBP2aB) gene, whereas the CBFβ subunit is encoded by the CBFB (PEBP2B) gene. The CBFα subunit binds directly to the DNA promoter sequences of target genes involved in hematopoiesis, whereas the beta subunit does not bind to DNA but enhances the DNA affinity of the CBF complex and protects it from proteolysis. t(8;21) targets the RUNX1 gene on chromosome 21q22 and the Eight twenty One (ETO) gene on chromosome 8q22.
RUNX proteins bind DNA as heterodimers through CBFβ, and both DNA-binding and heterodimerization are mediated through a central RUNX domain that has a high degree of homology with the Drosophila protein Runt. As described above, CBFβ does not directly contact DNA, but instead increases the stability and DNA-binding affinity of RUNX proteins. CBFs play a critical role in the development of several different cellular lineages. For example, RUNX1 is a master regulatory protein that controls the formation of definitive hematopoietic stem cells (HSCs) (3). Expression of RUNX1 and CBFB genes appears to be essential for the development of normal hematopoiesis. In murine knockout models, homozygous loss of Runx1 or CbFb results in embryonic death due to lack of definitive hematopoiesis (4). At the molecular level, chromosomal aberrations form RUNX1–RUNX1T1 and BCFB–Myosin, heavy chain 11, smooth muscle (MYH11) fusion genes, which disrupt subunits α and β of CBF. RUNX1–RUNX1T1 can be generated not only by t(8;21), but also by its infrequent variants, including complex translocations involving one or two additional chromosomes (5-8) and insertions [e.g. ins(21;8)(q22q22;q22) and ins(8;21)(q22;q22q22)] (5-7, 9), although these are more rare. CBF–AML represents one of the most common cytogenetic abnormalities in acute myeloid leukemia (10), comprising of 4-12% of newly-diagnosed cases of AML (11). Patients with CBF-AML have a relatively favorable outcome, with a significantly lower median age and better prognosis than patients with AML associated with a normal karyotype or chromosomal aberrations. The favorable outcome, with higher complete remission (CR) rate and lower incidence of relapse, especially for patients who receive high-dose cytarabine in post-remission treatment (12-14), has led to the formation of several collaborations aimed at discussing the indications for allogeneic stem cell transplantation (SCT) in CBF-AML (15-17).
Unlike the cytogenetic definition of CBF-AML, patient outcome does not appear to be as homogeneous as its cytogenetic definition. Indeed, 30-50% of patients with CBF-AML experience relapse and the 5-year survival is only 50% (18). The prognosis of patients with CBF-AML is heterogeneous, and is frequently complicated by additional chromosomal or molecular aberrations. Specifically, additional chromosomal aberrations are detected at diagnosis in 40% of patients with inv(16)/t (16;16) and in 70% of t(8;21) patients (2, 19). Several studies have suggested that secondary chromosome aberrations may have clinical significance, and that there is also a difference between patients with inv(16)/t(16;16) and those with t(8;21). Schlenk et al. reported that secondary aberrations such as +8, +21, and +22 are more common in those with inv(16)/t(16;16), while -Y,-X, and del(9q) are more common in those with t(8;21) (17). However, most of these additional chromosomal aberrations fail to exhibit clinical significance. For example, male patients with t(8;21) and -Y have a short overall survival and duration of first CR (17). On the contrary, a study from China showed that male patients with t(8;21) and -Y have a good prognosis (11). Several acquired genetic mutations and changes in both gene and microRNA expression that occur in addition to t(8;21)(q22;q22) and inv(16)(p13q22)/t(16;16)(p13;q22), the cytogenetic hallmarks of CBF-AML, have been recently reported. The most common mutations are receptor tyrosine kinase (RTK) c-KIT mutations and Fms-like tyrosine kinase 3 (FLT3) mutations, which are present in 12-47% of patients with t(8;21), and in 22-38% of those with inv(16)/t(16;16). Haploinsufficiency of the putative tumor suppressor genes Transducer-like enhancer of split-1 (TLE1) and Transducer-like enhancer of split-1 (TLE4) in patients with t(8;21) with del(9q), Meningioma 1 (MN1) overexpression in patients with inv(16), and epigenetic and post-transcriptional silencing of CCAAT/enhancer-binding protein alpha (CEBPA) have also been reported (2). Rat sarcoma (RAS) mutations have been identified in 36% of inv(16)/t(16;16) and 5% of t(8;21) cases. In a model proposed by Gilland et al., AML was suggested to result from class-I mutations conferring a proliferative advantage to AML and class-II mutations leading to impaired hematopoietic differentiation. With respect to CBF-AML, the c-KIT, FLT3, and RAS genes may be sites of class I mutations, while RUNX1–ETO and CBFβ–MYH11 gene fusions represent class-II mutations. Several other studies have shown that c-KIT mutations are associated with shorter event-free survival and overall survival. Especially for t(8;21) AML, KIT mutations occur mostly in exon 17 and confer an adverse prognosis (20-25), whereas the prognostic significance of KIT mutations (both in exons 8 and 17) in inv(16) AML is not well-established. KIT mutations can be targeted by tyrosine kinase inhibitors, which are selectively active against specific KIT mutations. For instance, imatinib is active against various exon 8 mutations and the exon 17 mutation involving codon N822, but not against mutations involving codon D816, which can be successfully targeted with dasatinib and midostaurin (26).
The mutation status of RAS is not associated with patient prognosis, and thus the clinical relevance of RAS mutations remains controversial. Among patients with inv(16)/t(16;16) and t(8;21) AML, clinical outcomes are heterogeneous, which is contrary to the cytogenetic definition. This observation may be related to various gene aberrations, additional chromosomal abnormalities, and specific clinical features. For these reasons, it is important to consider clear standards on the basis not only of chromosomal differences, but also of comprehensive prognostic factor analysis including gene aberration and clinical features. Thus, the aims of the present study were to analyze genetic and clinical markers in patients with CBF-AML patients and to identify patients who require intensive treatment.
Patients and Methods
Between January 2000 to December 2010, 657 patients were diagnosed with AML. We identified the patients with t(8;21) AML and the patients with inv(16)/t(16;16) AML through successful cytogenetic analysis of bone marrow (BM). The following clinicopathological variables and treatment outcomes were retrospectively collected: Patient demographics, complete blood count, liver function test, renal function test, and C-reactive protein levels. We also collected information regarding additional chromosomal abnormalities and gene mutations via analysis of bone marrow or peripheral blood blast samples. The presence of FLT3-Internal-tandem duplications (ITD) was analyzed by Polymerase chain reaction (PCR) on genomic DNA using primer pair 11F and 12R. Detection of c-KIT mutations was performed using genomic DNA with PCR fragment analysis and direct sequencing. We only screened exon 17 D816 and N822 mutations, which were assayed together. To analyze the mutation status of exon 8 in c-KIT, we performed direct sequencing.
Treatment. Patients received induction therapy according to the following doses: cytarabine of 200 mg/m2 for seven days in combination with idarubicin of 12 mg/m2 for three days. As part of consolidation, patients received three to four cycles of high-dose cytarabine and idarubicin as follows: cytarabine of 3 g/m2 every 12 h on days 1, 3, and 5 in combination with idarubicine of 12 mg/m2 on days 2 and 4.
Statistical analysis. We analyzed prognostic factors related to overall survival, progression-free survival (PFS), and leukemia-free survival (LFS). For overall survival, events were defined as death from any cause. Overall survival was measured from the first date of chemotherapy to the date of death or the date of the last follow-up visit. CR was defined as recovery of morphologically-normal BM and normal blood counts (neutrophils ≥1500/μl and platelets ≥100,000/μl) and no circulating leukemic blasts or evidence of extramedullary leukemia. Relapse was defined by ≥5% BM blasts, circulating leukemic blasts, or development of extramedullary leukemia. Relapse-free survival was measured from the date of the first administration of chemotherapy to the date of relapse, death, or last follow-up visit.
Clinical and laboratory findings before treatment.
Leukemia-free survival was measured from the date of the first CR to the date of relapse, death, or last follow-up visit. Survival analysis was estimated using the Kaplan–Meier method and compared by the log-rank test. Multivariate survival analysis was carried out using a Cox regression model. Results that reached a level of p<0.05 were considered statistically significant. Estimates for hazard ratios and corresponding 95% confidence interval (CI) were obtained for each significant prognostic factor.
Results
Clinical features and treatment outcomes. Between January 2000 to December 2010, approximately 67 (10.2%) out of the 657 patients with newly-diagnosed of AML were found to have CBF-positive AML. We identified 51 (7.8%) patients with t(8;21) AML and 16 (2.4%) patients with inv(16)/t(16;16) AML through successful cytogenetic analysis of BM. The median age of patients was 44 (range: 18-75) years, and 31 (46.3%) patients were male. Clinical and laboratory findings obtained before starting treatment are summarized in Table I. Only hemoglobin levels were significantly different between patients with t(8;21) and patients with inv(16)/t(16;16). A total of seven patients were enrolled in the t(8;21) AML group, while only one patient was enrolled in the inv(16) AML group; the age of patients in both groups was >60 years. The overall CR rate was 92.3% (62/67), the median overall survival was 80.6 months and median relapse-free survival was 68.4 months.
We analyzed the relationship among pre-treatment clinical and laboratory findings and treatment outcomes including overall survival and leukemia-free survival. Univariate analysis indicated that thrombocytopenia (≤20×103/μl), a high percentage of blasts in peripheral blood (>50%), and a high percentage of blasts in BM (>50%) were all significantly associated with poor overall survival (Table II, Figure 1). However, sex, old age (>60 years), leukocytosis or leucopenia, and low hemoglobin levels were not significantly associated with survival. In leukemia-free survival, thrombocytopenia (≤20×103/μl) and high percentage of blasts in BM (>50%) were significantly associated with a shorter duration of leukemia-free survival, while patients with Y chromosome deletions exhibited longer leukemia-free survival (Table II). With respect to relapse-free survival, thrombocytopenia (≤20×103/μl) and high percentage of blasts in BM (>50%) were significantly associated with poor progression-free survival, while Y chromosome deletions were favorable factors for relapse-free survival. A total of 11 (21.6%) patients with t(8;21) AML and 5(31.3%) patients with inv(16)/t(16;16) AML received BM transplantations. Most patients who received BM transplantation had additional chromosomal abnormalities or c-KIT mutations. In our study two patients with multi-lineage dysplasia identified during an initial bone marrow study also experienced a relapse after 24 months and had a poor overall survival of less than 36 months. Ultimately, these two patients died from uncontrolled infections.
Clinical features and treatment outcomes.
Additional chromosomal abnormalities and gene mutations. A total of 25 (49.0%) patients with t(8;21) had additional chromosomal abnormalities, the most common of which were deletions of the sex chromosomes X or Y. Specifically, 15 of the 25 male patients with t(8;21) AML and one of the 6 male patients with inv(16) were found to have a Y chromosome deletion. In addition, two patients with a Y chromosome deletion also had a deletion of chromosome 9. The remaining chromosomal abnormalities were deletion of chromosome 9 or 7, complex chromosomal abnormality, and abnormal 19q 13. In patients with inv(16)/t(16;16) AML, only 6 (37.5%) patients had a secondary chromosomal abnormality, of which trisomy 21 was the most common (Table III).
There were two patients with multi-lineage dysplasia observed at the initial BM examination, both of whom had a poor prognosis. Conversely, male patients with Y chromosome deletions had a good prognosis with a hazard ratio of 0.39 (95% CI=1.15-9.14) and p-value of 0.02. Y chromosome deletions were also associated with short relapse-free survival (p-value=0.04). The statistical power of the Y chromosome deletion was maintained not only for cases of t(8;21) AML, but also for cases of total CBF-AML.
We next checked the c-KIT mutation status in 27 patients, and found that c-KIT mutations were present in 5 (18.5%) patients with CBF-AML. A total of two patients had exon 17 t(8;21) AML, one patient had a mutation in exon 8, and three patients had mutations in exons 10 and 11. However, in our study, c-KIT mutations were not clinically significant because of the small number of patients. We also analyzed the status of FLT3-ITD in 26 patients with CBF-AML, but did not identify any mutations.
We next performed a multivariate analysis of all of the variables that showed a significant effect on overall survival in univariate analysis including thrombocytopenia (≤20×103/μl), high percentage of blasts in the peripheral blood (>50%), high percentage of blasts in BM (>50%), and Y chromosome deletions. In male patients, thrombocytopenia (≤20×103/μl), high percentage of blasts in peripheral blood (>50%), high percentage of blasts in BM (>50%), and Y chromosome deletions retained their significance in multivariate analysis (Table IV). In female patients however, only thrombocytopenia (≤20×103/μl) and a high percentage of blasts in the peripheral blood (>50%) were associated with poor overall survival in multivariate analysis (Table IV).
Discussion
CBF–AML is defined by the presence of either t(8;21)(q22;q22) or inv(16)(p13;q22)/t(16;16)(p13;q22) chromosomal re-arrangements that result in the disruption of CBFA and CBFB genes encoding CBF α and β subunits, respectively. CBF-AML accounts for approximately 4-12% of all cases of AML. CBF-AML is associated with a significantly lower median age of patients, as well as improved prognosis compared to patients with AML with a normal karyotype or other chromosomal aberrations. The favorable outcome associated with CBF-AML is also associated with a higher rate of CR and a lower incidence of relapse, especially for patients receiving high-dose cytarabine in post-remission treatment, which has led to several collaborative efforts to discuss the indications of allogeneic SCT in CBF-AML. The clinical heterogeneity of CBF-AML is well known, with 30-50% of patients with CBF-AML experiencing relapse and a 5-year survival of only 50%. The heterogeneity of CBF-AML prognosis is likely complicated by additional chromosomal or molecular aberrations, and thus several attempts have been made to identify prognostic factors to identify aspects of pre-treatment clinical features and genetic findings (27). Previous studies on prognostic factor analysis can be divided into pre-treatment clinical features including ethnicity, sex, and laboratory findings; additional chromosomal abnormalities including +8, +21, +22, and deletion of sex chromosomes; estimation of minimal residual disease; and mutations of c-KIT, FLT3, and RAS. However, it has not yet been possible to build a risk stratification strategy based on existing data.
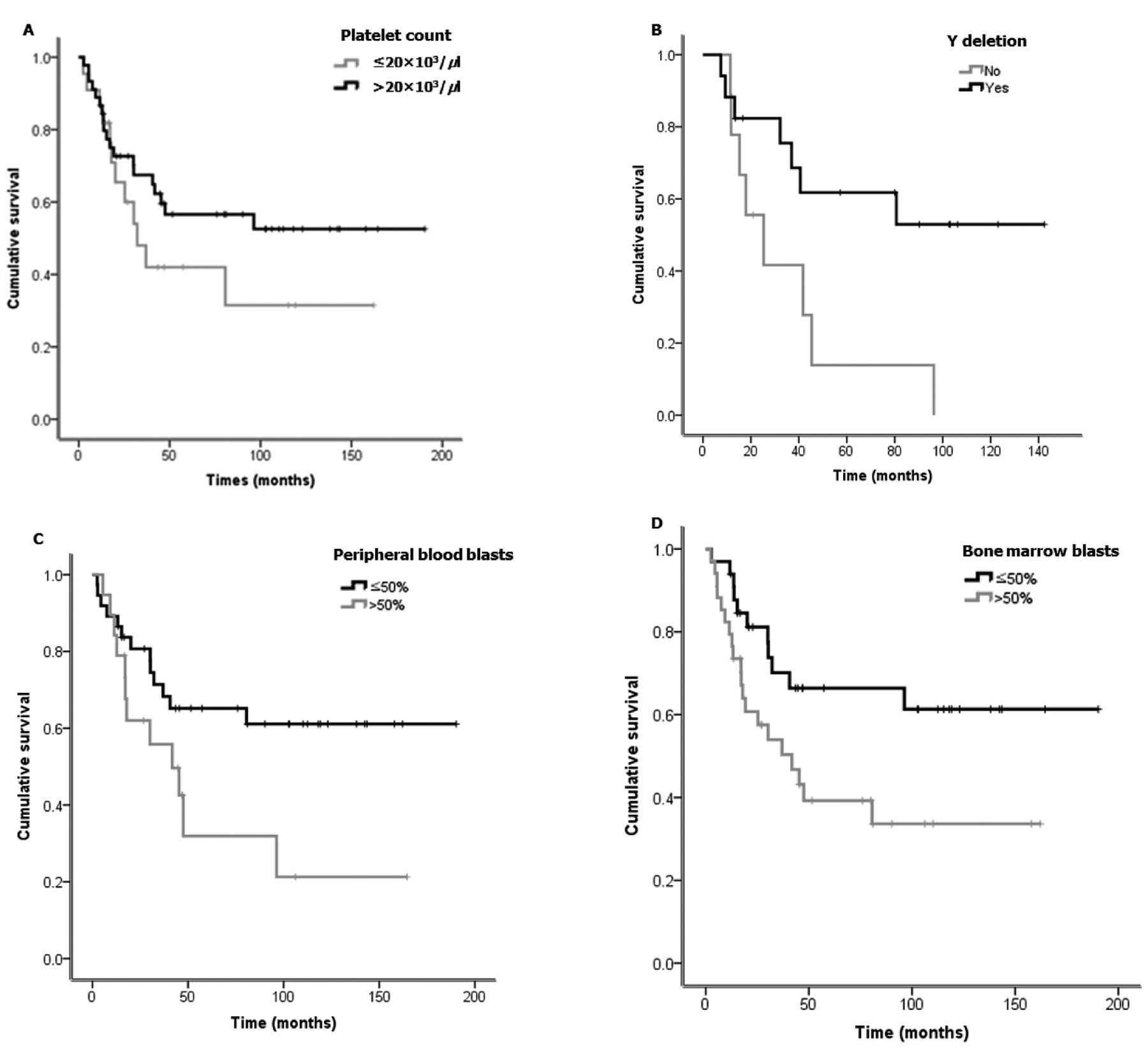
Survival curves based on the factors: platelet count (A), Y chromosome deletion (B), peripheral blood blasts (C), and bone marrow blasts (D).
The present study was designed to analyze all factors that might be related to treatment outcomes, including clinical, laboratory, and genetic factors in CBF-AML. Importantly, prognostic analysis in our study showed that deletion of the Y sex chromosome in male patients with CBF–AML was an independent indicator of favorable disease outcome, and may thus be considered as a favorable prognostic biomarker. According to multivariate analysis, thrombocytopenia (≤20×103/μl) and a high percentage of blasts in BM (>50%) were also associated with a poor prognosis.
Additional chromosomal abnormalities.
Multivariate analysis.
Several reports have discussed the clinical significance of additional sex chromosome abnormalities (28). Deletion of the Y chromosome is more common in children than in adults (29), and thus it is possible that there is an age-related heterogeneity of chromosomal aberrations and different genetic mechanisms between children and adults (30). In a previous study on additional sex chromosomal abnormalities in CBF-AML, the clinical significance of the results proved controversial. Specifically, Schelnk et al. reported that male patients with a Y chromosome deletion had significantly reduced overall survival and duration of first CR (17). However, in a study performed in children, additional chromosomal abnormalities, including sex chromosomes, were found to reflect relatively late stages of leukemia and their presence before treatment accurately predicted poor prognosis (31-33). Several other reports showed that the prognostic impacts of these additional changes were negligible (34-38). Consistent with the results of our study, several other reports have shown that male patients with Y chromosomal deletions have a good prognosis (39); however, most of these studies consisted of sub-group analyses of a small number of patients, and thus the clinical significance of Y deletions in CBF-AML has not yet been clearly defined.
In our study, two patients with multi-lineage dysplasia during an initial BM study experienced a relapse after 24 months and had a poor survival of less than 36 months. These patients died due to uncontrolled infection. According to a study by Ma et al., de novo AML involving multi-lineage dysplasia is associated with poor response to therapy, while dysplasia in t(8;21) AML has not been shown to influence prognosis (40). However, they analyzed dysplasia in hematopoietic lineages rather than multilineage dysplasia, and thus their results suggest that patients with multilineage dysplasia may exhibit increased vulnerability to infections, although this will require evaluation in a large-scale.
In the present study, we analyzed 67 patients with CBF-AML, taking into account the incidence of secondary genetic aberrations (c-KIT, FLT3) (2). However, the patient sample size was small, and thus our results did not appear clinically significant. c-KIT is a proto-oncogene that encodes a type-III trans-membrane tyrosine kinase that functions as a receptor for the cytokine stem cell factor, also known as mast cell growth factor. Mutations of c-KIT are common in CBF-AML, and are present in 12.7% to 48.1% of all cases of AML1-ETO leukemia (20). Mutations at Asp816 and Arg822 in the tyrosine kinase domain are the two most common mutation in t(8;21) AML (24), and several reports have shown that various c-KIT mutations adversely affect the frequency of relapse and overall survival of patients with CBF-AML (41, 42). Several c-KIT mutations are associated with poor treatment outcome, including mutations at position Asp816 in exon 17 and several mutations appearing in exon 8. In particular, the Asp816 mutation is found in 10.5% of all cases of t(8;21) AML and is associated with poor overall survival and a high rate of relapse.
Laboratory findings have implicated both leukocytosis and thrombocytopenia in influencing CBF-AML outcome. Several additional reports have shown that low platelet counts may reflect poor treatment outcomes (18, 37). Similarly, Billstrom et al. showed that the presence of leukocytosis at the initial diagnosis of CBF-AML is associated with a less favorable prognosis (18, 38, 40, 43). In the present study, however, neither leukocytosis nor leucopenia appeared to be related to treatment outcome.
A limitation of the present study was the gene mutation test, which was performed for only a few patients and thus had very limited statistical power. However, according to a previous study (44), of a total of 116 patients, mutKIT17 and mutKIT8 were identified in 36 (31%) and seven (6%) patients, respectively. In patients with t(8;21), prognosis was significantly worse in patients with mutKIT17 compared to those without the mutation. Interestingly, this difference was limited to adults. In patients with inv(16), there was no prognostic impact of c-KIT mutations, and therefore an analysis of mutKIT17 in adult CBF–AML patients with t(8;21) is recommended for a predictive prognosis (44). The incidence of aberrant cluster of differentiation-56 (CD56) expression was significantly higher in patients with a mutation of c-KIT in exon 17 compared to those without a mutation (20-21).
FLT3 is a class-III receptor tyrosine kinase in a family that also includes KIT. FLT3 is expressed in both early hematopoietic stem cells and in a subset of dendritic cell progenitors. FLT3 signaling activates intracellular pathways [e.g. RAS-Rapidly Accelerated Fibrosarcoma (Raf)-mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (Mek), phosphatidylinositol 3-kinase (PI3K)]-v-akt murine thymoma viral oncogene homolog 1provided (AKT) that promote proliferation and inhibition of apoptosis. The most commonly described FLT3 mutation in AML is the ITD mutation of the juxtamembrane segment. The FLT3-ITD mutation leads to loss of the autoinhibition exerted by the juxtamembrane domain over the tyrosine kinase domain, generating a constitutively active FLT3 molecule. FLT3-ITD mutations are found in 20% to 30% of patients with AML and are more common in normal karyotype AML, acute promyelocytic leukemia, and AML with a t(6;9)(p23;q34) translocation (9-14). Patients who have FLT3-ITD-positive normal-karyotype AML have a higher leukocyte count and exhibit a rate of complete response similar to that of FLT3-ITD-negative patients, but have shorter disease-free survival and overall survival, mainly because of frequent relapses (45-50). We did not observe any FLT3-ITD mutations in the patients analyzed in this study.
The results of our study are meaningful because of our decision to collect and analyze all factors that might be related with treatment outcomes including clinical, laboratory and genetic factors in CBF-AML in South Korea. Indeed, previous studies have only analyzed a few factors. Additionally, the heterogeneity of CBF-AML may be related to ethnicity and age, and this is the first such prognostic factor analysis of CMF-AML in Korea.
Conclusion
The results of the present study suggest that deletion of the Y chromosome may be a favorable prognostic factor for patients with CBF-AML, while thrombocytopenia (≤20×103/μl) and a high percentage of blasts in BM (>50%) were associated with a poor prognosis. Further studies are required to confirm the feasibility of risk stratification for use in risk-adapted therapy.
Footnotes
-
↵* These Authors contributed equally to this work.
- Received December 9, 2013.
- Revision received January 3, 2014.
- Accepted January 7, 2014.
- Copyright© 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
Jump to section
Related Articles
Cited By...
- Functional Properties of KIT Mutations Are Associated with Differential Clinical Outcomes and Response to Targeted Therapeutics in CBF Acute Myeloid Leukemia
- Clinical Impact of Additional Cytogenetic Aberrations, cKIT and RAS Mutations, and Treatment Elements in Pediatric t(8;21)-AML: Results From an International Retrospective Study by the International Berlin-Frankfurt-Munster Study Group