


Abstract
Background/Aim: Tumor angiogenesis induced by vascular endothelial growth factor (VEGF) and/or fibroblast growth factor (FGF) plays an important role in tumor growth, metastasis, and drug resistance. However, the characteristics of tumor vessels derived from these angiogenic factors have not been fully explored. Materials and Methods: To functionally examine tumor vessels, we developed in vivo VEGF- and FGF-induced tumor blood vessel models. We performed immunohistochemistry and Hoechst perfusion assay to elucidate histopathological differences between the derived tumor vessels. To kinetically understand tumor perfusion, we employed radiolabeled PEGylated liposomes. Results: While tumor vessel density was substantially increased by enhanced expression levels of VEGF and FGF, permeability of VEGF-driven tumor vessels was significantly higher than that of FGF-driven ones, the latter demonstrating an increased number of pericyte-covered vessels. Accordingly, we observed an increased tumor retention of the PEGylated liposomes in the VEGF-driven tumor. Conclusion: Our in vivo models of tumor vessel demonstrate the frequency of pericyte coverage and tumor perfusion levels as major functional differences between VEGF- and FGF-driven tumor vessels.
- Angiogenesis
- vascular endothelial growth factor
- fibroblast growth factor
- immunohistochemistry
- permeability
- pericytes
Angiogenesis, the formation of new blood vessels from pre-existing blood vessels, plays critical roles in tumor development and metastasis (1-4). In most aggressive types of cancer, angiogenic factors have been widely reported as promising targets for therapeutic intervention (5, 6). Specifically, vascular endothelial growth factor (VEGF), which was initially described as a vascular permeability factor because of its ability to induce vascular leakage (7), has been considered as the most important key regulator of angiogenesis in various cancer types (8-10). Thus, the field of angiogenesis research has progressed rapidly with advances in the evaluation of angiogenic growth factors led by VEGF, which specifically regulates endothelial cell proliferation (11-13). In accordance with research, blocking VEGF signaling has shown significant efficacy in preclinical tumor models and human patients with cancer. However, although clinical antitumor effects with short-term tumor control benefits are often observed, anti-VEGF therapies seldom prevent relapse to progressive disease (2). Moreover, the mechanism of angiogenesis has not been sufficiently elucidated because it is a complex process involving activation, migration, invasion, proliferation, and tube formation of vascular endothelial cells.
It has been reported that several angiogenic factors, besides VEGF, are related to these complicated mechanisms of angiogenesis. Fibroblast growth factor (FGF) is another well-known angiogenic factor which is stored in the vascular basement membrane (14), and FGF signaling pathways also have been implicated in vascular development and progression (15). In normal tissues, while VEGF-induced blood vessels contain high numbers of endothelial fenestrations that mediate high permeability, FGF-induced blood vessels lack vascular fenestrations (16). In recent years, FGF signaling pathways have been suggested to contribute to tumor angiogenesis, cell proliferation, and resistance to chemotherapy and anti-VEGF therapy for several types of human cancer in preclinical and clinical studies (17-20). However, these reports were inconclusive and insufficient for analyzing the differences between angiogenesis induced by VEGF and that by FGF in tumors. It is important to distinguish VEGF-driven and FGF-driven tumor vessels for proper selection of anticancer treatment in the clinical setting. Therefore, elucidating the characteristics of FGF-induced tumor angiogenesis that are distinct from VEGF-induced vessels could potentially be advantageous to improve current therapies for patients with cancer.
Unique structural features of abnormal angiogenesis, including hypervasculature and defective vascular structure, are observed in tumors. Enhanced vascular permeability caused by defective vascular structure in tumors is also a distinctive characteristic and mechanism, and is known as the enhanced permeability and retention (EPR) effect, a well-characterized concept by which molecules of certain sizes tend to accumulate in tumor tissue (21-23). The biodistribution and pharmacokinetics of PEGylated liposomes have been evaluated clinically to measure permeability and retention of tumor blood vessels noninvasively in patients with cancer (24, 25).
In this study, we engineered pancreatic cancer (KP-1) cells that develop tumor vessels with either VEGF or FGF dependency in vivo, and examined the characteristics of these two types of tumor vessels by histopathological analysis, as well as functional analysis using radiolabeled liposomes. After the histopathological analysis of VEGF- and FGF-induced tumor vessels, we verified these results using long-term circulating PEGylated liposomes as validated tools for performing leakage analysis of mouse tumor in vivo (26).
Materials and Methods
Drugs. Gemcitabine hydrochloride was purchased from Eli Lilly (Indianapolis, IN, USA), dissolved in distilled water, and stored at −20°C at a stock concentration of 20 mM. Sorafenib tosylate was purchased from Bayer (Tokyo, Japan) and dissolved in dimethyl sulfoxide (DMSO) (Wako, Osaka, Japan) at a stock concentration of 20 mM for in vitro use.
Establishment and culture of cell lines. To evaluate the effects of the angiogenic factors VEGF and FGF, we developed VEGF- and FGF-overexpressing cell lines. Human pancreatic cancer cell line KP-1, which released small amounts of angiogenic factors originally (27), was stably transfected with plasmids expressing human VEGF-A121 or mouse FGF4, as well as mock plasmid driven by human cytomegalovirus promoter as described in our previous research article (28) to produce KP-1/VEGF, KP-1/FGF, and KP-1/mock cells. All cell types were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), penicillin (100 units/ml), and streptomycin (100 μg/ml) in all experiments.
Cell growth assay. The number of viable cells was determined by quantifying cellular ATP, which was measured using CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Southampton, UK). Signal intensity was determined using luminometer (Perkin Elmer, Norwalk, CT, USA). To examine cell growth of the pancreatic cell line, all four types of KP-1 cells were seeded at 500 cells per well of a 96-well plate. At 5 min, 24 h, 48 h, 72 h, and 168 h after cell seeding without drug treatment, the cell number was evaluated. The relative growth rate was defined as the number of cells relative to that at 5 min. In the drug treatment experiments, the four cell lines were seeded at a density of 1,000 cells per well in 96-well culture plates and incubated in RPMI-1640 with 10% FBS overnight. On the following day, the cells were treated with serially diluted series of gemcitabine or sorafenib. The controls received DMSO (vehicle) at a concentration equal to that of the sorafenib-treated cells. The cell growth-inhibitory effects of gemcitabine or sorafenib were examined at 72 h after treatment.
Quantification of endothelial growth factors by enzyme-linked immunosorbent assay (ELISA). KP-1/VEGF, KP-1/FGF, and KP-1/mock cells (1×106 cells/well, 100 mm dishes) were cultured with complete growth medium for 48 h. The culture medium was then changed to serum-free medium and culture supernatants were collected 24 h later. VEGF and FGF secreted into the medium from the cells were diluted and measured using ELISA kits [human VEGF: SEA143Hu (detection range: 15.6-1,000 pg/ml), mouse FGF: SEA034Mu (detection range: 7.8-500 pg/ml); USCN Life Science, Wuhan, China].
Animals. All animal experiments were conducted according to the Guidelines for Animal Experiments approved by both Eisai Institutional Animal Care and Use Committee, and Committee for Ethics of Animal Experimentation at National Cancer Center. The approval numbers are 13-C-0063, 13-C-0117, and 13-C-0213. Female CAnN.Cg-Foxn1nu/CrlCrlj mice (6 weeks old) were purchased from Charles River Laboratories Japan (Yokohama, Japan). Five mice were housed per plastic cage with paper chip bedding in an air-conditioned animal room maintained at 23±2°C and 60±5% relative humidity with 12 h light/dark cycles. Basal diet (CE-2; CLEA, Tokyo, Japan) and water were available ad libitum throughout the experiment.
Subcutaneous xenograft models. KP-1/VEGF, KP-1/FGF, and KP-1/mock cells (8×106 cells/inoculation) were inoculated subcutaneously into the right flanks of 7 week-old mice. KP-1/mock was used as a negative control. Approximately 3-5 weeks after transplantation, all experiments were performed using mice bearing tumor sized at approximately 500 mm3. Tumor size was measured using a caliper. Tumor volume was calculated by the formula: tumor volume (mm3)=length (mm) × [width (mm)]2 × 0.5 (29, 30). For the sorafenib treatment in mice, sorafenib tosylate was dissolved in Cremophor EL/ethanol [50:50; Cremophor EL (Sigma, St. Louis, MO, USA), 99.5% ethanol (Wako, Osaka, Japan)] as 4-fold (4×) stock solution, and diluted to 1× solution with distilled water before administration. Sorafenib at 30 mg/kg was orally administered once daily for 8 days.
Histopathological evaluation of vascular permeability. To investigate tumor perfusion and microvessel density (MVD) induced by VEGF or FGF, we performed Hoechst 33342 perfusion assay and immunohistochemical (IHC) staining using anti-CD31 antibody. The tumor-bearing mice were intravenously injected with 10 mg/ml (0.1 ml/mouse) Hoechst 33342 (Invitrogen, Waltham, MA, USA), a marker of tumor perfusion, and were sacrificed 5 min after the injection. The tumor tissue was resected from each mouse, embedded in OCT compound (Sakura Finetek, Tokyo, Japan) and frozen on dry ice. Frozen tissue blocks were sectioned at 10 μm. Tumor vascular perfusion was evaluated by randomly measuring 16 Hoechst 33342-positive areas using four of each harvested tumor cryosections for the xenografted tumors (31).
For quantitative analysis of MVD, IHC staining with anti-CD31, a marker of blood vessels, was performed. Tumor sections were fixed with acetone at −20°C and then blocked by phosphate-buffered saline (PBS) containing 1% bovine serum albumin. The tissue sections were then incubated with 1:200-diluted phycoerythrin (PE) anti-mouse CD31/platelet endothelial cell adhesion molecule 1 (PECAM-1) antibody (clone MEC 13.3, 553373; BD Pharmingen, San Jose, CA, USA) overnight at 4°C. After washing with PBS three times, the slides were mounted with ProLong® Gold Antifade Reagent (Cell Signaling Technology, Beverly, MA, USA). Four dense regions with Hoechst 33342 fluorescence signals from four tumor sections (n=4 mice for each tumor type) were captured as representative regions using a fluorescence microscope (BZ-9000; KEYENCE, Osaka, Japan) with a 20× objective lens. Furthermore, the CD31 fluorescence signals were also captured from each representative region. The fluorescence signals of Hoechst 33342 and CD31 in the captured images were quantified using Lumina Vision (ver. 2.2.2; Mitani Corporation, Fukui, Japan), and averages of the perfusion levels and vessel number were calculated. To evaluate functional blood vessels, CD31-positive vessels associated with Hoechst 33342-positive cells were counted in 16 objective fields from four tumor sections (n=4 mice for each tumor type) under the microscope at 200× magnification, and the index of functional vessels was calculated using the formula:

Immunohistopathological examination of vascular structure. To address the differences in permeability level among VEGF- and FGF-derived tumor blood vessels, vascular structure was analyzed by employing double staining with CD31 (a marker for blood vessel) and α-smooth muscle actin (α-SMA) (a marker for pericytes). The tumors were fixed in IHC zinc fixative (BD Biosciences, San Jose, CA, USA) for 24 h and embedded in paraffin. Zinc-fixed paraffin-embedded tumor tissues were used for identification of CD31/PECAM-1 and α-SMA staining. The tumor samples (n=4 for each tumor type) were sectioned at 4 μm, mounted on slides, and air-dried for 30 min. Sections were de-paraffinized and rehydrated in PBS, and endogenous peroxidase activity was blocked in 3% hydrogen peroxide solution. After PBS rinses, the sections were incubated with rat anti-mouse antibody to CD31 (clone MEC 13.3, 550274; BD Pharmingen, San Jose, CA, USA) or rabbit anti-mouse antibody to α-SMA (ab5694; Abcam, Cambridge, MA, USA) overnight at 4°C, and then incubated for 30 min at room temperature with Histofine® Simple Stain Mouse MAX PO (for rat) and Histofine® Simple Stain AP (for rabbit) (Nichirei Bioscience, Tokyo, Japan). After the incubation with secondary antibody, brown color for anti-rat secondary antibody was developed with incubation in 3,3’-diaminobenzidine solution for 10 min at room temperature, and red color for anti-rabbit secondary antibody stained with new fuchsin for 10 min at room temperature. The sections were rinsed with distilled water, counterstained with Mayer's hematoxylin for 1 min. The pixels of CD31 signals in the captured images were quantified using Adobe Photoshop Elements software (ver. 2.0; Adobe Systems, San Jose, CA, USA) as previously described (32), and average vascular numbers were calculated. All CD31-positive vessels associated with α-SMA-positive cells were counted in 16 objective fields from four tumor sections (n=4 mice for each tumor type) at 200× magnification, and the index for pericyte-covered vessels was calculated using the formula:

Preparation of 111In-diethylenetriaminepentaacetic acid (DTPA) encapsulated PEGylated liposomes. PEGylated liposomes were prepared by following the lipid film hydration extrusion method as described previously (33). The lipid mixture containing distearoylphosphatidylcholine (NOF Corporation, Tokyo, Japan), 1,2-distearoyl-phosphatidylethanolamine-methyl-polyethyleneglycol conjugate-2000 (Genzyme, Baar, Switzerland), and cholesterol (NOF Corporation) at a molar ratio of 60:6:34 were dissolved in chloroform, and the solvent lipids were deposited as a thin film in an egg-plant shaped flask by rotary evaporation. After removal of any residual solvents, the film was hydrated by adding 30 mM HEPES buffer (pH 7.4) containing 5% mannitol and 10 mM DTPA at 60°C, and then repeatedly extruded through 0.2-, 0.2-, and 0.1-μm pore-sized polycarbonate membrane filters (GE Healthcare, Buckinghamshire, UK) to formulate liposomes at diameter of approximately 120 nm. Free DTPA was removed by passing through a Sephadex G-50 fine (GE Healthcare Japan) gel filtration. Phospholipid concentration was measured using phospholipid assay kit (Wako, Osaka, Japan). 111In-DTPA encapsulated PEGylated liposomes were generated by following the remote loading method; specifically, the liposomes encapsulating DTPA were labeled with 111In (Nihon Medi-Physics, Tokyo, Japan) through incubation with 111In-oxine (34).
Distribution of 111In-DTPA encapsulated PEGylated liposomes in living KP-1 tumor mouse xenograft models. In order to kinetically understand the different characteristics of VEGF- and FGF-driven tumor vessels, especially tumor permeability, we utilized a validated method that we previously developed to quantitatively measure tumor perfusion using radiolabeled liposomes (35). 111In-DTPA encapsulated PEGylated liposomes (approximately 500 kBq/2 μmol phospholipids/0.2 ml saline) were injected intravenously into the mice bearing each type of KP-1 tumor, sized at approximately 500 mm3 (n=5 for each tumor type). We collected tumors, principal organs, and blood at 5 min, 24 h, and 72 h after the liposome administration, and measured radioactivity using a gamma counter (PerkinElmer, Hopkinton, MA, USA).
Statistical analysis. Data are expressed as the mean±SD. We used the statistical software GraphPad Prism software ver. 7.02 (GraphPad Software, La Jolla, CA, USA). Dunnett's multiple comparison test was used in the statistical analysis among the cells or vessels induced by each growth factor. An unpaired t-test was used in the analysis between vehicle-control group and sorafenib-treated group. Differences with p-values lower than 0.05 were considered statistically significant.
Results
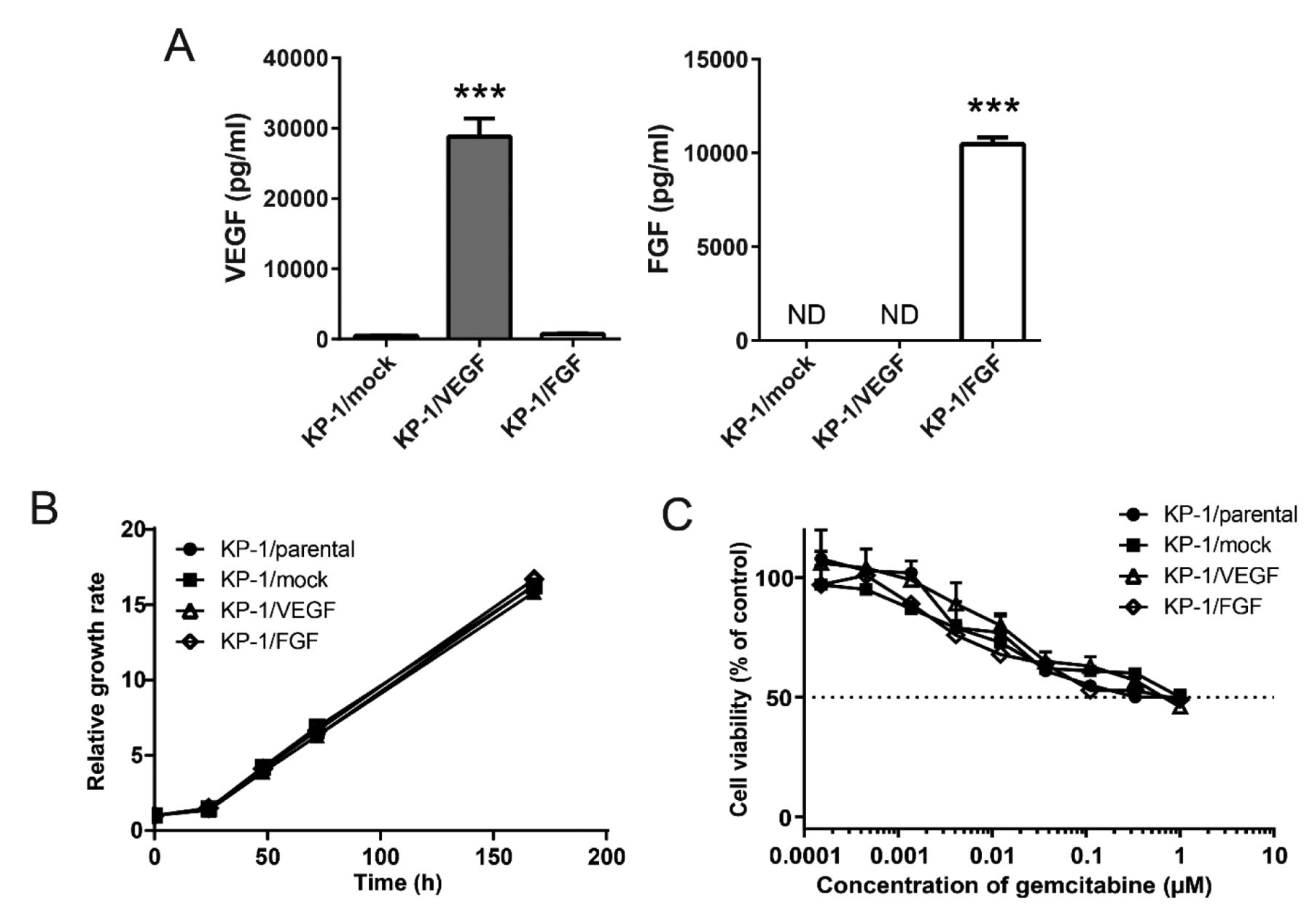
Establishment of VEGF and FGF overexpressing pancreatic tumors cell lines. To confirm that angiogenic factors were properly expressed, we measured angiogenesis factors secreted from our KP-1 transfectants by ELISA. We confirmed that the levels of secreted VEGF, and of FGF were significantly increased in the correspondingly transfected cells compared to the cells transfected with control vector (Figure 1A).

Development of vascular endothelial growth factor (KP-1/VEGF)- and fibroblast growth factor (KP-1/FGF)-overexpressing pancreatic cell lines. A: Amounts of VEGF and FGF proteins in the cell culture supernatants. The amounts were determined by enzyme-linked immunosorbent assay kits. Data are presented as the mean±SD. ***Significantly different at p<0.001 compared to KP-1/mock cells. B: Relative growth rate of induced tumor cells in vitro. The numbers of cells were measured by quantifying the ATP level. C: Sensitivity of KP-1/VEGF and KP-1/FGF to gemcitabine in in vitro cell culture. After 72 h-treatment, the numbers of cells were measured by quantifying the ATP level. ND: Not detected.
Since stable transfection can potentially induce abnormal genetic alteration and affect cell growth and drug sensitivity, we examined the growth rates of these cells in vitro. We confirmed that there was no significant difference in cell growth among the parental (KP-1 cells) and the transfected KP-1/VEGF, KP-1/FGF, and KP-1/mock cells (Figure 1B). We also observed similar growth inhibition patterns under gemcitabine treatment, which is commonly used to treat pancreatic cancer (Figure 1C). These results indicate that the genes we inserted did not modify other properties of these cancer cells, including cell growth and intrinsic sensitivity to this cytotoxic drug.
Different characteristics of the tumor blood vessels induced by VEGF and FGF in in vivo tumors. Since angiogenic factors play an important role in tumor proliferation via alteration of its microenvironment, we examined the growth speed of the tumors in vivo. Both KP-1/VEGF and KP-1/FGF tumors grew more rapidly than KP-1/mock tumor; in particular, the speed of growth of KP-1/FGF cells was significantly higher than that of the other types (Figure 2A).
To examine the sensitivity to anti-VEGF therapy of tumor blood vessels in the xenografted tumors in vivo, we evaluated the anti-angiogenic activity of sorafenib. Firstly, we evaluated the sensitivity to sorafenib in vitro and similar growth inhibition was observed among all cell types (Figure 2B). We next assessed the sensitivity in vivo in order to appropriately understand the tumor microenvironment using these models. We reasoned that if blood vessel formation in the tumor in vivo was dependent on VEGF, then sorafenib treatment would reduce tumor formation only in tumors derived from KP-1 cells overexpressing VEGF. As expected, sorafenib (30 mg/kg) showed antitumor effect against KP-1/VEGF tumor, but not KP-1/FGF tumor (Figure 2C). Furthermore, we confirmed that sorafenib reduced the numbers of VEGF-driven but not FGF-driven blood vessels by IHC staining (Figure 2D-F). These findings indicate that the tumor blood vessels in these tumors were functionally dependent on the respective angiogenic factor, and these angiogenesis models are useful for analyzing the differences between VEGF- and FGF-regulated tumor vessels.

Dependency on angiogenesis factors in constructing tumor vessels in in vivo tumor models. A: The relative growth rate of in vivo xenografted vascular endothelial growth factor (KP-1/VEGF)- and fibroblast growth factor (KP-1/FGF)-overexpressing tumors. Data are presented as the mean±SD. ***Significantly different at p<0.001. B. The sensitivity to sorafenib in in vitro cell culture. After 72 h-treatment, the numbers of cells were measured by quantifying the ATP levels. C: Antitumor activity of sorafenib against KP-1/VEG and KP-1/FGF subcutaneous tumor in nude mice. Sorafenib (30 mg/kg) was orally administered for 8 days to five mice xenografted with tumors. Data are presented as the mean relative tumor volume±SD. **Significantly different at p<0.01 compared to vehicle-control group. ▴: Treatments. D, E: Histopathological findings of CD31-positive tumor vessels pre- (D) and post- (E) sorafenib treatment. After 8-day treatment, we sacrificed mice, and the tumors were collected. KP-1/mock tumors were collected as a control. Collected tumors were embedded in paraffin and immunostained with anti-CD31 (brown color). Scale bar=100 μm; magnification, ×200. F: The mean microvessel density as shown by staining with anti-CD31 at ×200 magnification. Data were analyzed by unpaired t-test. ***Significantly different at p<0.001. NS: Not significantly different.

Histopathological findings of the permeability of tumor vessels developed in KP-1/mock, vascular endothelial growth factor (KP-1/VEGF)- and fibroblast growth factor (KP-1/FGF)-overexpressing tumors. A: Representative images of collected tumors double-stained with CD31 and Hoechst 33342. The structure of tumor vessels was stained with anti-CD31 (red color) to detect angiogenesis in the frozen sections of KP-1/mock, KP-1/VEGF, and KP-1/FGF tumors. The blood vessel permeability was assessed by the Hoechst 33342 (blue color) fluorescent area. Scale bar=400 μm; magnification, ×100. B: The mean area of permeability stained with Hoechst 33342. C: The mean proportion of functional vessels that were double-stained with anti-CD31 and Hoechst 33342 at ×100 magnification views. D: The permeability per functional vessel in each tumor is shown. All data are presented as the mean±SD. Data were analyzed by Dunnett's test. Significantly different at *p<0.05, **p<0.01 and ***p<0.001. NS: Not significantly different.
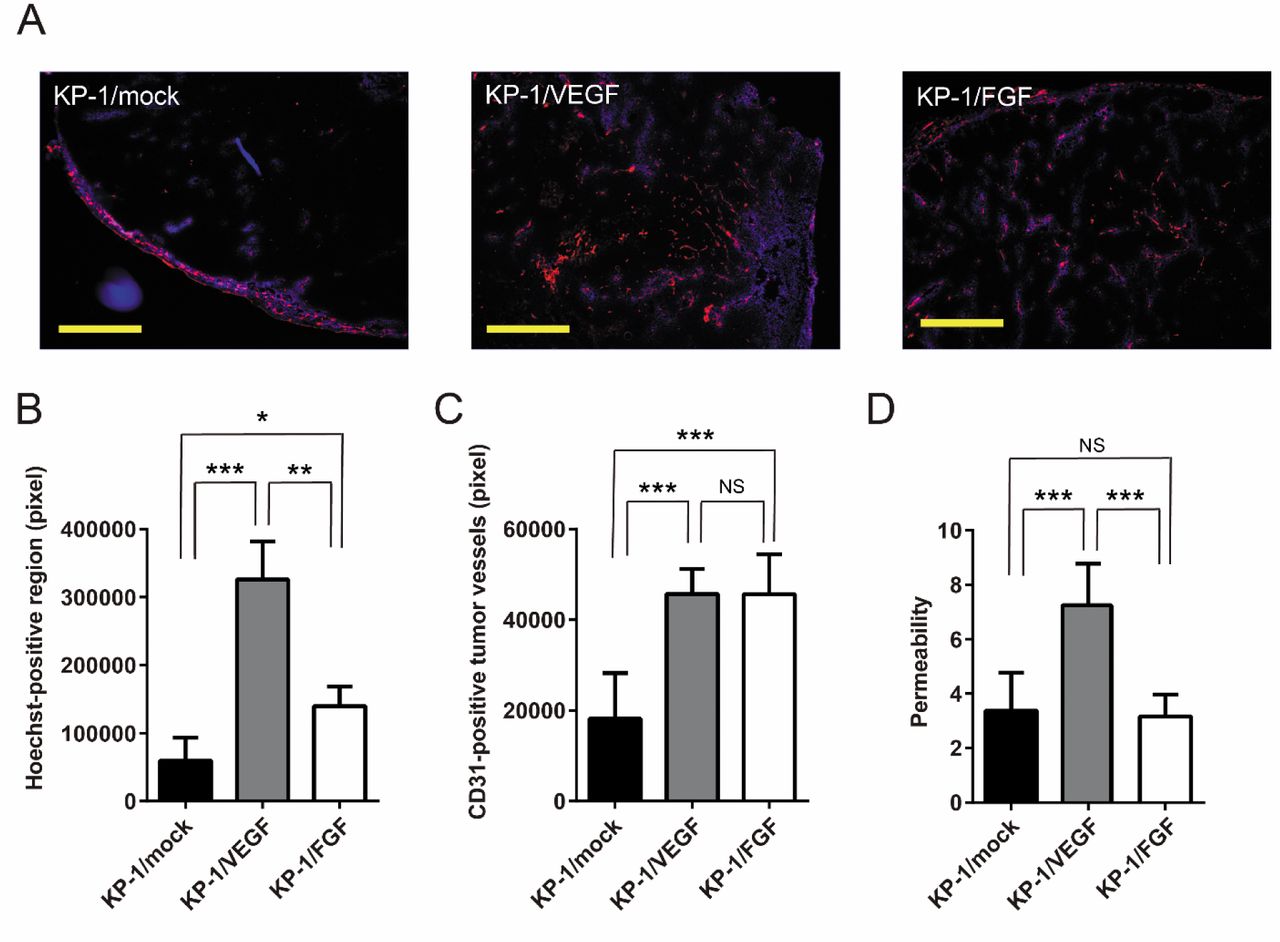
Histopathological analysis of tumors blood vessels. IHC data obtained from Hoechst 33342 perfusion and staining by anti-CD31 showed that the MVD was significantly increased in KP-1/VEGF (3.5-fold) and KP-1/FGF (2.4-fold) xenograft tumors compared to the control tumor (KP-1/mock) (Figure 3A). The permeability of blood vessels, which was defined as Hoechst 33342-positive regions, in the KP-1/VEGF and KP-1/FGF tumors were 5.5- and 2.4-fold larger than that of the control tumor (Figure 3B). Regarding Hoechst-positive functional blood vessels, which have adequate blood flow, 2.5-fold increases in both KP-1/VEGF and KP-1/FGF tumors were observed when compared to the KP-1/mock xenografts (Figure 3C). However, 15%, 39%, and 12% of the blood vessels in the KP-1/mock, KP-1/VEGF, and KP-1/FGF tumors, respectively, were not stained with Hoechst 33342. Consequently, these vessels were defined as non-functional vasculature (data not shown). The ratio of permeability per functional vessel in KP-1/VEGF tumor was 2.2-fold larger than that of KP-1/mock and KP-1/FGF, which indicated that VEGF-induced tumor vessels were leakier than FGF-induced vessels (Figure 3D).

Histopathological findings of the structure of tumor vessels developed in KP-1/mock, vascular endothelial growth factor (KP-1/VEGF)- and fibroblast growth factor (KP-1/FGF)-overexpressing tumors. A: Representative images of collected tumors double-stained for CD31 and α-smooth muscle actin (α-SMA). The structure of tumor vessels was stained with anti-CD31 (brown color) to detect angiogenesis in KP-1/mock, KP-1/VEGF and KP-1/FGF tumors. The pericyte coverage around tumor vessels was detected by α-SMA (red color). Scale bar=100 μm; magnification, ×200. B: The mean ratio of the pericyte-covered vessels to all vessels. All data are presented as the mean±SD. Data were analyzed by Dunnett's test. Significantly different at *p<0.05 and ***p<0.001.
The FGF-driven vessels had high pericyte coverage. IHC data obtained from staining by anti-CD31 and anti-α-SMA showed that vessels in the KP-1/FGF and KP-1/mock tumors were highly covered by pericytes (>75%). On the other hand, only 26.7% of tumor vessels were covered by pericytes in the KP-1/VEGF xenografted tumor (Figure 4). These findings imply that permeability in the tumor were dependent on pericyte coverage in the tumor.
The distribution of 111In-DTPA-encapsulated PEGylated liposomes in the KP-1 tumor mouse xenograft models. We evaluated the biodistribution of 111In-DTPA PEGylated liposomes in the KP-1/VEGF, KP-1/FGF, and KP-1/mock xenograft models. Consistent with increased tumor vessels induced by VEGF and FGF, the accumulation of liposomes was significantly higher at 24 h after injection of the liposomes in both KP-1/VEGF and KP-1/FGF tumors than KP-1/mock tumors. By comparing KP-1/VEGF and KP-1/FGF tumors, significant difference in liposomal accumulation was observed at 72 h, wherein only KP-1/VEGF tumors retained a similar level of liposomal accumulation from the earlier time point (Figure 5A). There were no differences regarding the accumulation of liposomes in blood (Figure 5B) and other major organs (Figure 5C). These findings imply that increased MVD was crucial to the delivery of radiolabeled liposomes into the tumor, and leaky vessels contributed to retention of the liposomes in the tumor.
Discussion
It is crucial to develop unique in vivo models for understanding biological functions of angiogenesis by concurrently comparing each molecule under physiological conditions. However, few studies have compared, in vivo tumor vessel models for investigating the role of each angiogenic factor (36). In this study, we successfully established in vivo VEGF- and FGF-driven tumor blood vessel models by manufacturing cancer cells in accordance with each angiogenic factor. Since KP-1 cells seldom express angiogenic factors, our strategy allowed comparison of different characteristics associated with VEGF-driven and FGF-driven tumor vessels without considering the involvement of other angiogenic factors. By applying our experimental strategy to other potent angiogenic factors, we can expect rapid progress in exploring functions of angiopoietin, hepatocyte growth factor, platelet-derived growth factor, matrix metalloproteinase, etc. (37-40). These angiogenic factors are also known to be relevant for resistance to VEGF-targeted therapy (41-44). Inclusion of other angiogenic factors to further elucidate these vascular characteristics is important for the development of anti-angiogenic agents to overcome resistance to existing anti-angiogenic therapies.
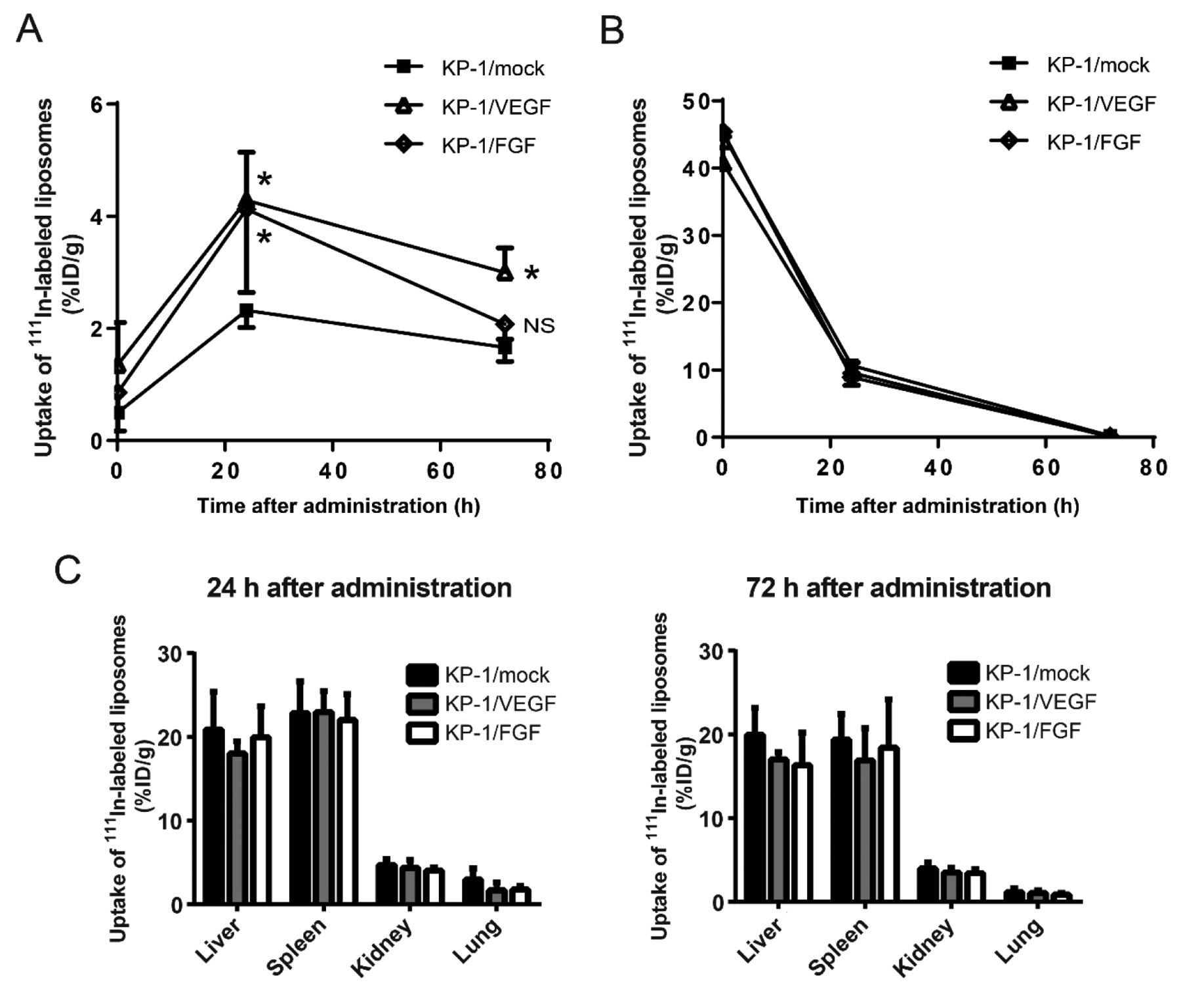
The ex vivo bio-accumulation of 111In-diethylenetriaminepentaacetic acid (DTPA) PEGylated liposomes at periodic points in KP-1/mock, vascular endothelial growth factor (KP-1/VEGF)- and fibroblast growth factor (KP-1/FGF)-overexpressing tumors. A: Five female mice bearing tumor of approximately 500 mm3 were injected with radiolabeled liposomes. At 5 min, 24 h, and 72 h after the treatment, the mice were sacrificed, and tumors and organs were collected. After sampling at 72 h, the radioactivity of all samples was evaluated using a gamma counter. All data are presented as the mean±SD. Data were analyzed by Dunnett's test. *Significantly different at p<0.05 compared with KP-1/mock tumor at each time point. NS: Not significantly different. B: Radioactivity from circulating liposomes in the blood is shown at 24 and 72 h after administration. C: Radioactivity from circulating liposomes in major organs is shown at 24 and 72 h after administration.
Interestingly, KP-1/VEGF cells and KP-1/FGF cells showed similar tumor MVD in the tumor xenografts, but the growth rate of KP-1/FGF tumors was significantly higher than that of KP-1/VEGF in vivo. These findings suggest that abnormal leakiness of tumor vessels may affect the growth of tumors, supplying insufficient amounts of oxygen and nutrition (45, 46). On the other hand, the vascular network generated by FGF-induced vessels may allow tumors to access adequate supply of oxygen and nutrients and removal of waste products; consequently, the tumors can grow steadily with a high growth rate. However, the abnormal leakiness of VEGF-induced tumor vessels might be useful to deliver anticancer drugs with large molecular size into tumors effectively due to the increase of endothelial fenestrations with low pericyte coverage. Indeed, our study indicated an increased extravasation and retention of liposomes by EPR effects in the VEGF-induced tumor blood vessel model. Therefore, the delivery of liposomal drugs based on EPR effects may be effective for tumors with high VEGF expression.
Since radiolabeled liposomes are highly quantitative tools for evaluating the amount of blood flow to tumors in vivo (47), we employed it to distinguish VEGF- and FGF-induced tumor vessels in this study. The results show that the liposomes leaked from abnormal vasculature, and the leaked liposomes were highly accumulated only in the VEGF-regulated tumor tissues. It follows from these findings that the comparison of the penetration of liposomes between VEGF- and FGF-driven tumor vessel models may provide much useful information in analyzing EPR effects. In addition, such an approach using radiolabeled liposomes might be a feasible method for characterizing the tumor vasculature and identifying tumors with higher accumulation of drugs non-invasively. However, further experiments in the preclinical and clinical settings to the adequate size of liposomes, and correlation analysis of liposomal accumulation and antitumor activity of liposomal drugs are warranted.
In summary, we established VEGF- and FGF-driven tumor blood vessel models reflecting angiogenesis induced by each factor accurately, and clearly demonstrated the different characteristics of VEGF- and FGF-driven tumor vessels in terms of vessel structure with pericyte coverage and drug distribution pattern. These features were correlated with resistance to anti-VEGF therapy of the tumors. Our findings provide novel research platforms that could be applicable for the development of new angiogenesis inhibitors through evaluating functions of other angiogenic factors, and provide mechanistic insights into drug resistance to anti-VEGF therapy.
Acknowledgements
The human pancreatic cancer cell line KP-1 was obtained from Dr. Akihiro Funakoshi of the National Kyushu Cancer Center. The authors would like to thank Dr. Yasuhiro Funahashi and Dr. Masao Iwata for their helpful advice in this article. This work was partially supported by Grant-in-Aid for Young Scientists B (S.H.: 26861034), Grant-in-Aid for Scientific Research B (I.O.U., H.F.: 24390297, 15H04911), and Grant-in-Aid for Challenging Exploratory Research (I.O.U.: 25670546) from Japan Society for the Promotion of Science. This work was also financially supported by Eisai Co., Ltd. under the provisions of the collaborative contract between the National Cancer Center and Eisai Co., Ltd.
Footnotes
This article is freely accessible online.
- Received September 6, 2017.
- Revision received September 25, 2017.
- Accepted September 27, 2017.
- Copyright© 2017, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}