


Abstract
In the past two decades, significant advances have been made in our understanding of colorectal (CRC) tumors with DNA mismatch (MMR) repair deficiency. The knowledge from molecular and genetic alterations in a variety of clinical conditions has refined the disease terminology and classification. Hereditary non-polyposis colorectal cancer (HNPCC) encompasses a spectrum of conditions that have significant phenotypic overlapping that makes clinical diagnosis a challenging task. Distinguishing among the HNPCC disorders is clinically important, as the approach to surveillance for patients and their at-risk family members differs according to risks for colonic and extracolonic cancer associated with each syndrome. Prospective and next-generation studies will provide valuable clinical information regarding the natural history of disease that will help differentiate the Lynch syndrome mimics and guide diagnosis and management for heterogeneous conditions currently grouped under the category of familial CRC. The review is intended to present and discuss the molecular nature of various conditions related to MMR deficiency and discusses the tools and strategies that have been used in detecting these conditions.
- 4ereditary non-polyposis colorectal cancer
- Lynch syndrome
- Lynch-like syndrome
- FCCTX
- mismatch repair
- review
We have entered a genomic era of cancer research which may lead to new possibilities of cancer treatment. Next-generation DNA sequencing has greatly improved the detection of DNA variants in the genome of each individual and contributes to a personalized management of an individual's cancer (1).
Colorectal cancer (CRC) is the third most common cancer and the fourth most common cause of cancer death in the world (2). Heredity represents a major cause of CRC, with up to 30% of the cases estimated to develop due to genetic factors and about 5% linked to inherited mutations in cancer-predisposing genes (3). Identifying these high-risk patients is a major issue because morbidity and mortality from CRC and extracolonic cancer in patients and their relatives can be reduced by early and intensive screening (4, 5).
Familial CRC was first described in 1966 by Lynch, who referred to type I for families with CRC only, and type II for families with cancer also including gynecological cancer (6). Later, the term hereditary non-polyposis colorectal cancer (HNPCC) was introduced to emphasize the lack of a polyposis phenotype. In 1984, the term ‘Lynch syndrome’ was proposed to describe this condition and has been most commonly used since then (7). Although the term HNPCC is often used interchangeably with Lynch syndrome, it is important to remember that HNPCC is a clinical diagnosis for patients and families that meet Amsterdam I or II criteria (AC), whereas diagnosis of Lynch syndrome requires presence of a genetically confirmed disease-predisposing mismatch repair (MMR) mutation (8, 9).
In order to stratify families for genetic analysis, the AC were developed to permit the identification of MMR defects and their association within tumor spectrum (10, 11). The AC requires at least three affected family members in two or more generations, with one being a first-degree relative of the other two and at least one individual diagnosed before 50 years of age (10, 12). AC-I applies to families with three or more cases of CRC and AC-II also includes extracolonic tumors, i.e. endometrial cancer, cancer of the upper urinary tract and cancer of the small bowel (10, 12). The Bethesda guidelines included the tumor marker of microsatellite instability (MSI), and the revised Bethesda criteria specified all cancer known at the time to be associated with the syndrome (11, 13).
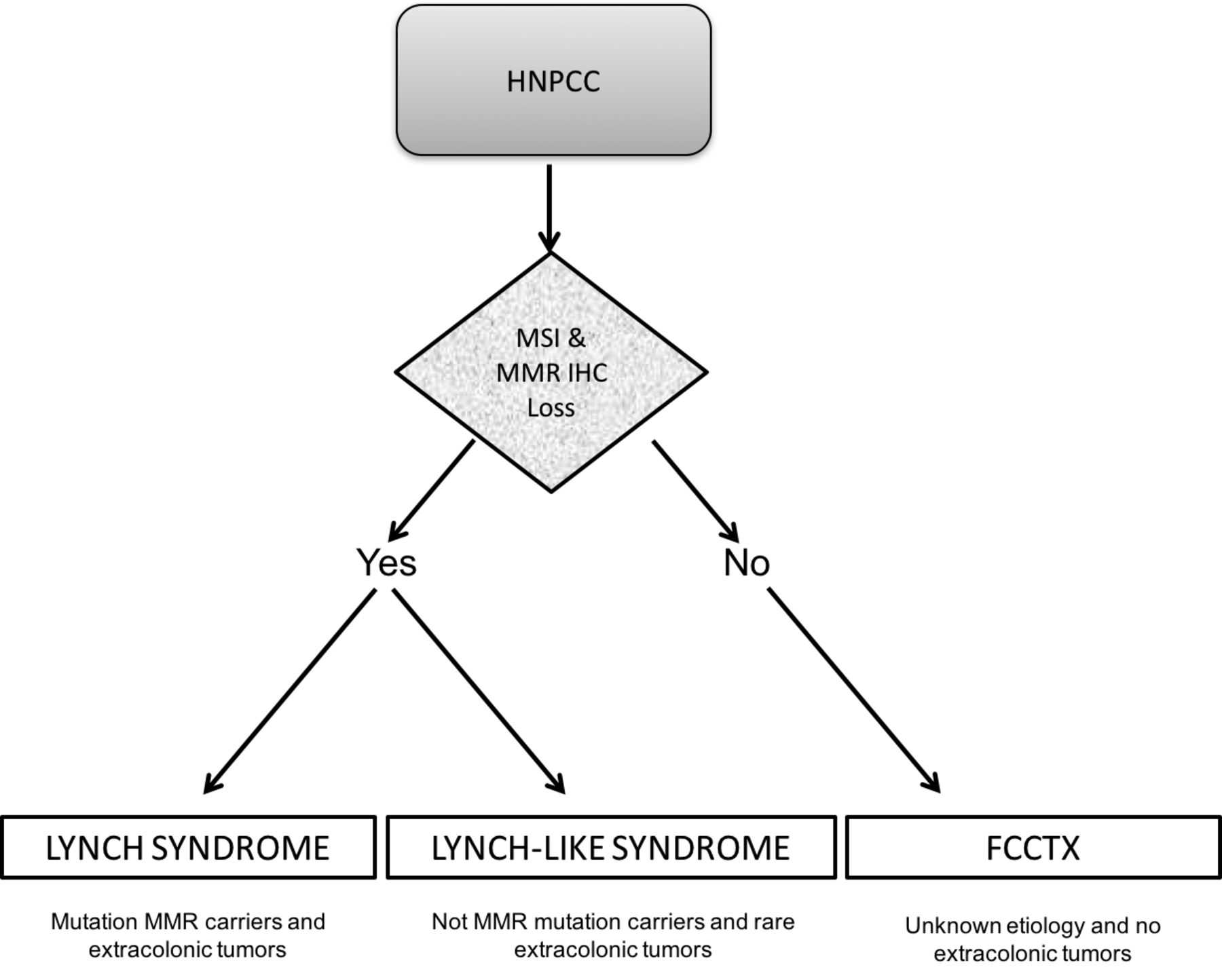
Among AC-positive families, disease-predisposing mutations in MMR genes characteristic of Lynch syndrome can be identified in ~60%, whereas 40% do not show MMR defects and are, accordingly, referred to as familial colorectal cancer type X (FCCTX) (14, 15). The term FCCTX was coined by Lindor and colleagues in 2005 to describe families that meet AC-I, but have MMR-proficient tumors. The polyp burden and risk factors of FCCTX have not been fully and independently characterized. Lynch-like syndrome describes cases where molecular testing demonstrates the presence of MSI with/without abnormal expression of MMR proteins on immunohistochemical (IHC) testing of tumors, without the presence of characteristic germline mutations seen in Lynch syndrome (16, 17) (Figure 1).
In this review, we discuss the recent knowledge on the molecular nature of familial CRC, and definitions of the various conditions related to MMR deficiency. The review aims to improve our understanding of hereditary CRC risk, pathogenesis and prevention.
Lynch Syndrome
Lynch syndrome is the most common inheritable type of CRC, accounting for 2% to 4% of CRC, and has an estimated prevalence in the general population of one in 440 (18-20). Lynch syndrome increases the risk for CRC (lifetime risk=70-80%), and endometrial (50-60%), stomach (13-19%) and ovarian (9-14%) cancer, and of the small intestine, biliary tract, and brain, as well as carcinoma of the ureters and renal pelvis (21). Nowadays, most patients with Lynch syndrome have been identified following investigation because of their family or personal history of multiple or early-onset cancer (4).
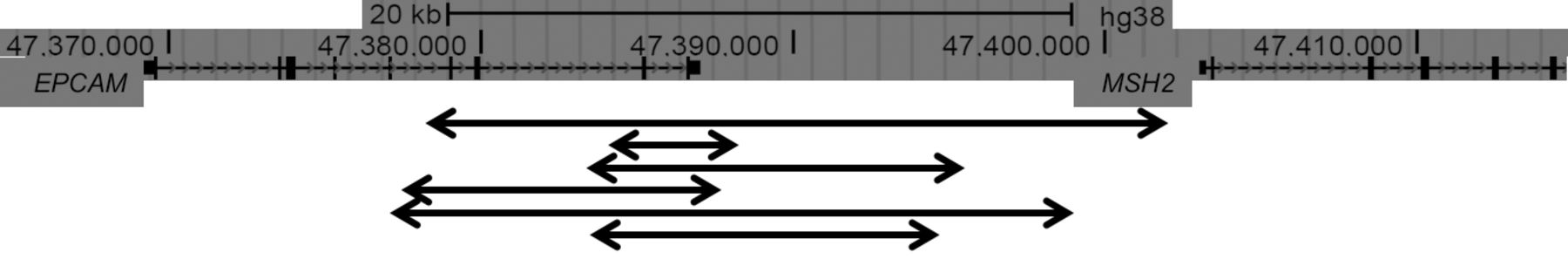
Lynch syndrome is caused by a defective MMR system due to the presence of germline defects in at least one of the MMR genes - mutL homolog 1 (MLH1), mutS homolog 2 (MSH2), mutS homolog 6 (MSH6) and postmeiotic segregation increased 2 (PMS2). An estimated 80-90% of Lynch syndrome is attributable to deleterious mutations in MLH1 and MSH2, with the remaining 10-20% due to mutations in MSH6 and PMS2. Moreover, up to 3% of Lynch syndrome is due to mutations in the epithelial cell adhesion molecule (EPCAM), which is directly upstream of MSH2. Deletions of the 3’-end of EPCAM result in epigenetic hypermethylation of the MSH2 promoter, producing a phenotype very similar to Lynch syndrome (9). Recent studies have described large deletions encompassing EPCAM exons 5 to 9 and others, including a large deletion involving both EPCAM and MSH2 genes (22-26) (Figure 2). The cumulative incidence of any cancer at 70 years of age is 72% for MLH1 and MSH2 mutation carriers but lower in MSH6 (52%) and PMS2 (18%) mutation carriers. MSH6 and PMS2 carriers developed no cancer before 40 years of age (4).
To date over 3,100 unique DNA variants across the MMR genes have been described in the International Society for Gastrointestinal Hereditary Tumors (InSIGHT) database (http://insight-group.org/variants/database/) and a recent clinical InSiGHT consensus classification identified 57% of the MMR variants as pathogenic or likely pathogenic (Class 5 and 4), 32% as uncertain variants (Class 3), 4% as likely not pathogenic (Class 2) and 7% as not pathogenic (Class 1) (27).
Lynch-like Syndrome
The term Lynch-like syndrome, or suspected Lynch syndrome has been suggested to describe patients with tumors showing deficient DNA MMR expression, but no identified deleterious germline mutation in MMR genes (Figure 1). B-Raf proto-oncogene, serine/threonine kinase (BRAF) V600E mutation or MLH1 promoter hypermethylation have not been found in Lynch-like tumors, demonstrating that the deficiency of MLH1 expression is not due to BRAF mutation (16, 17). Lynch-like syndrome may account for up to 70% of cases in which Lynch syndrome is clinically suspected with a high MSI profile and absence of a MMR proteins (17).
From a clinical perspective, patients with Lynch-like syndrome present cancer at younger ages, similarly to those with Lynch syndrome (17). Buchanan et al. described family histories of Lynch-like syndrome patients with high incidence of metachronous and synchronous CRC and fulfillment of AC (28). On the other hand, Mas-Moya and colleagues identified significant differences between patients with Lynch-like syndrome and those with Lynch syndrome (29). The majority of those with Lynch-like syndrome had CRC in the right colon (93%) when compared to those with Lynch syndrome (45%). In this regard, out of all patients with left-sided or rectal adenocarcinoma, 96% (23/24) demonstrated germline mutations (defined as Lynch syndrome) (29).
There are growing hypothesis regarding the pathogenesis of Lynch-like syndrome (28, 30, 31). Some authors suggested that patients with Lynch-like syndrome could actually have Lynch syndrome but their germline mutations in MMR genes might not be detectable by current testing (28). Recently, Liu and colleagues identified a patient with family history of early-onset CRC carrying an unbalanced paracentric inversion spanning exon 2 to 6 of MSH2 (30). In this regard, Rhees et al. found a significant proportion of patients with previously unexplained MSH2-type Lynch syndrome harboring an inversion from exon 1 to 7 in MSH2 gene (31).

Hereditary non-polyposis colorectal cancer (HNPCC) as a familial colorectal cancer aggregation. At least three different entities based on immunohistochemistry (IHC) and microsatellite instability (MSI) analysis have been proposed, including Lynch syndrome, Lynch-like syndrome and familial colorectal cancer type X (FCCTX). MMR: Mismatch repair genes (MMR).
There are other mechanisms that inactivate DNA MMR genes which could result in tumors that resemble Lynch syndrome. For instance, around 60% of Lynch-like CRCs exhibit bi-allelic somatic inactivation of MMR genes within the tumor (32-34). A somatic mutation in one allele of MMR genes along with loss of heterozygosity of the other allele is the most common described pattern (17, 35). These somatic MMR gene mutations are likely sporadic events, suggesting that such tumors are most likely cancers with sporadic DNA MMR deficiency.
Familial Colorectal Cancer Type X
Families meeting AC-I for Lynch syndrome, but not carrying deleterious alterations in MMR genes, nor MSI, are defined as having FCCTX (36), however, some studies have also included AC-II with microsatellite stable (MSS) tumors (36-40). CRCs in FCCTX families are diagnosed at a slightly older age compared to those with Lynch syndrome and the risk of extracolonic cancer is not more than that of the average-risk population (14).
The FCCTX subset is challenging, not least since the clinical presentation and the histopathological features mimic sporadic MMR-proficient tumors. The genetic causes are unknown but candidate genes include e.g. centromere protein E (CENPE), kinesin family member 24 (KIF24), polypeptide N-acetylgalactosaminyltransferase 12 (GALNT12), zinc finger protein 367 (ZNF367), hyaluronan binding protein 4 (HABP4), gamma-aminobutyric acid type B receptor subunit 2 (GABBR2) and bone morphogenetic protein 4 (BMP4). Differences in genomic and gene-expression profiles do exist, e.g. gain of chromosome 20q, global hypomethylation and up-regulation of the G-protein-coupled receptor-signaling pathway (40).
Alterations in APC, WNT signaling pathway regulator (APC) and KRAS proto-oncogene, GTPase (KRAS)/Raf-1 proto-oncogene, serine/threonine kinase (RAF1) signaling pathway have been described in FCCTX, resembling those in Lynch syndrome and sporadic CRC, respectively (41-43). In addition, it has been suggested that base-excision repair pathways and epigenetic events could be implicated in FCCTX pathogenesis (41, 42).
A subgroup of FCCTX tumors related to the CpG island methylator phenotype pathway have been characterized by simultaneous methylation of gene promoters resulting in transcriptional silencing, high MSI and BRAF mutations, as well as a subgroup arising through the chromosomal instability pathway, characterized by aneuploidy and LOH, which is thought to be involved in most CRC (43).
The clinical phenotype of FCCTX substantially differs from that Lynch syndrome families, which includes fewer cases of CRC, higher age of CRC onset, more tumors of the left colon, and fewer extracolonic manifestations (43-46). According to Kravochuck and Church, families with FCCTX do not have a specific syndrome but may have one of several genotypes, including mutY DNA glycosylase (MUTYH)-associated polyposis, nth-like DNA glycosylase 1 (NTHL1)-associated polyposis, polymerase proofreading-associated polyposis, or serrated polyposis (35).
The overall risk of cancer is lower in FCCTX than in Lynch syndrome (14, 47), in which the lifetime CRC risk is estimated to be 50-80%. Age at diagnosis of CRC differs, with a mean age of 48 years in Lynch syndrome and 60 years in FCCTX (14, 48-51). Synchronous and metachronous adenomatous polyps are more common in FCCTX than in Lynch syndrome (46). Further studies are required, however, since tumors arising within FCCTX families also appear to have a different pathological phenotype, with fewer tumor-infiltrating lymphocytes than those from families with Lynch syndrome (46).
Extended and in-depth analyses of the FCCTX tumor genome, methylome and proteome in well-defined tumor series could shed light on the basic mechanisms for potential application in refined diagnosis and targeted interventions (40).
Recommendations for Surveillance
Hereditary CRC syndromes provide a wealth of opportunities for highly targeted clinical management and prevention (52, 53). The key to prevention is early diagnosis through a comprehensive family history, followed by germline mutation testing if appropriate, and targeted surveillance and management for patients with mutations (52, 53).
For Lynch syndrome, initiating colonoscopy is recommended at age 20-25 years (or the age of 30 and 35 years in patients with MSH6 and PMS2 mutations, respectively) or 2-5 years prior to the earliest case of CRC, if it is diagnosed before the age of 25 years, and repeated every 1-2 years (9, 53). No specific upper limit is established and this should be based on the individual's health status (54, 55). Endometrial and ovarian cancer screening may be performed on a yearly basis from age 30-35 years with gynecological examination, pelvic ultrasound, cancer antigen 125 (CA125) analysis, and endometrial aspiration biopsy.
The American Society of Clinical Oncology endorsement panel concurs with the European society for medical oncology recommendations (55) and the National comprehensive Cancer Network guidelines (https://www.nccn.org/professionals/physician_gls/f_guidelines.asp#genetics_colon) for Lynch syndrome that prophylactic removal of the uterus and ovaries might be an option in female carriers from age 35 years and after childbearing is completed (56). Surveillance for other Lynch-associated cancers is recommended based on family history and may include upper endoscopy, and abdominal ultrasound with urine cytology from age 30-35 years at 1-2 year intervals. In Lynch syndrome, there is an increased risk of synchronous and metachronous CRC, with a risk of 16% of developing a second CRC after 10 years of follow-up (54, 55). Therefore, the need for intensive surveillance after surgery versus the option of an extended colectomy should be discussed at the time of CRC diagnosis, especially for young patients. Although some preclinical data suggest that MMR status may play a role as factor predictive of chemosensitivity, as well as prognosis and treatment, current evidence does not allow definitive recommendation on chemotherapy regimens to be made based on MSI status (54).
Surveillance programs in FCCTX are targeted at CRC and the mean age at onset of 60 years implies that surveillance colonoscopies are generally recommended with 3- to 5-year intervals, starting 5-10 years before the earliest age at onset in the family (44, 54). CRC surveillance in FCCTX families is very important. More efforts are needed to improve the identification of patients with familial CRC since a modestly increased risk of CRC has been described for these families (14, 54).
In terms of chemoprevention for CRC, there are reasonably compelling data supporting that aspirin is protective against CRC and some extracolonic tumors (57). Although current guidelines do not routinely recommend its use, recent data from the Colorectal Adenoma/Carcinoma Prevention Program (CAPP2) in a randomized, placebo-controlled trial showed a significant 60% reduction in the incidence of CRC and other Lynch syndrome-associated cancers among those using 600 mg of aspirin per day for at least 2 years (55, 58).
Understanding the molecular genetic mechanisms of these hereditary cancer subsets is relevant in order the able to discriminate between high- and low-risk groups, to identify novel predisposition loci, and improve targeted cancer control measures and pharmacological therapy. The lessons from hereditary cancer have dual clinical implications related to genetic counseling and diagnostics for hereditary cancer and insights into pathways and prognostic markers in CRC in general. Knowledge of familial CRC risks has the potential to impact patient management and subsequent screening and surveillance strategies. There is a need for preventive strategies that can utilize biomarkers in order to stratify patients into appropriate screening or surveillance programs.
Footnotes
This article is freely accessible online.
- Received July 1, 2016.
- Revision received July 14, 2016.
- Accepted July 18, 2016.
- Copyright© 2016 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Significance of the Multi-gene Panel myRisk in Japan
- Curative and Prophylactic Surgery of Young-onset Colorectal Cancer in Inherited Syndromes: A 15-Year Monocentric Retrospective Experience
- Frequency of Mismatch Repair Protein Deficiency in a Puerto Rican Population with Colonic Adenoma and Adenocarcinoma
- Schizosaccharomyces pombe MutS{alpha} and MutL{alpha} Maintain Stability of Tetra-Nucleotide Repeats and Msh3 of Hepta-Nucleotide Repeats
- MicroRNAs in the etiology of colorectal cancer: pathways and clinical implications