


Abstract
Optic pathway glioma (OPG) is a rare neoplasm and a defining feature of neurofibromatosis type 1 (NF1), a tumor suppressor genetic disorder. OPG predominantly arises during childhood. In contrast to sporadic OPG, this neoplasm frequently appears to show a more favorable course. Outcome appears to depend on localization of tumor; however, the correlation of imaging findings and visual acuity is in general low. Treatment for symptomatic OPG is not well standardized. Furthermore, determination of visual acuity as the most important parameter of follow-up control is often difficult to determine, particularly in children. Focal abnormal signal intensity (FASI) is a characteristic finding on magnetic resonance imaging (MRI) of NF1 patients. The aim of this study was to evaluate clinical and imaging findings of NF1 patients affected with OPG. Patients and Methods: Data of 925 NF1 patients with appropriate MRI cranial sectional images (N=1,948) were evaluated. A further 50 patients with cranial computed tomograms were included in the study. We compared imaging and clinical findings with respect to localization of OPG. Furthermore, we compared follow-up in treated individuals to those who were only regularly re-examined. The presence of FASI on MRI was determined and correlated to the occurrence of OPG. Dodge classification was applied to categorize OPG location. Results: OPG was diagnosed in 134 patients. The mean age of patients with symptomatic OPG was 7.6 years (n=57, 42.5%) and 11.6 years (n=77, 57.5%) in asymptomatic patients. The female to male ratio was about 1.1:1. In 48 symptomatic patients, the findings of initial ophthalmological investigations were available. In symptomatic patients, reduced visual acuity was the predominant finding. Strabismus (25%), exophthalmos (22.9%) and amblyopia (20.8%) were most frequently noticed, followed by endrocrinological abnormalities (14.6%). However, these findings did not differ between patients who were treated or who were subjected to a ‘wait-and-see’ policy. We could not verify an effect of therapy on vision in patients treated for OPG compared to symptomatic patients without treatment. OPG affecting the total optic pathway was more frequently diagnosed in symptomatic patients. FASI did not correlate with functional OPG status. Conclusion: OPG in NF1 is symptomatic in slightly less than 50% of affected individuals. This neurological finding may show a wide range of symptoms. At present, no established treatment protocol emerges from the history of the patients of this study and also from the literature. Although the onset of symptomatic OPG is strongly associated with early childhood, late onset of symptomatic OPG is a feature of adult NF1. Research for association of FASI to neurological findings in these patients should be based on other issues than association with OPG.
- Neurofibromatosis type 1
- optic pathway glioma
- brain tumor
- magnetic resonance imaging
- orbital tumor
- visual acuity
- focal areas of signal intensity
- brain tumor
- skull base
- skull base tumor
Optic pathway gliomas (OPG) are a characteristic finding in Neurofibromatosis type 1 (NF1). The prognosis of OPG in NF1 is estimated to be good in general, but a thorough investigation of affected individuals in many cases reveals substantial health risks that are evidently associated with this peculiar condition. Relatively few alternative therapies are available for OPG. This report describes experience in the diagnosis, treatment and follow-up of NF1 patients affected with OPG, based on a detailed analysis of the pertinent literature.
Characteristics of NF1. NF1 (Recklinghausen's neurofibromatosis) is a disease inherited in an autosomal dominant manner that is characterized as a multi-organ illness belonging to the evolving class of tumor suppressor syndromes (1-11). The incidence is estimated to be 1:3,000 (9) to 1:2,500 (12) live births. Penetrance is almost complete in affected individuals, but the phenotype varies considerably (13-16).
Neurofibromas are benign tumors of the nerve sheath and the hallmark of and eponymous for the disease (17). Pilocytic astrocytoma (PA), plexiform neurofibroma (PNF), certain osseous dysplasia and vasculopathies arise much more rarely in NF1 patients (18), but can cause more severe diseases than the prominent nodule-like cutaneous neurofibroma. However, these numerous NF1-associated multi-organ findings underline the fragmentariness of filing NF1 exclusively in the neurocutaneous syndromes or ‘phakomatoses’ (19). Mean life expectancy in NF1 is about 8 years below average (8). Malignant peripheral nerve sheath tumors (MPNST) are a defining complication in the course of NF1-associated diseases, which is one reason for reduced life expectancy (10). MPNST of the orbit in NF1 is a very rare finding. Cerebral tumors comprise the large spectrum of diseases noted in NF1 (20). With respect to OPG in NF1, the presence of orbital and/or ophthalmic plexiform neurofibroma can have a severe impact on visual acuity, irrespective of the potential additional impact of a co-existent OPG on optic nerve function.
The NF1 gene is localized on chromosome 17q11.2 and codes for the protein neurofibromin. The functions of neurofibromin are to a large extent unknown, but in several functions the protein acts as a tumor suppressor (21). The NF1 gene probably shows one of the highest mutation rates in the human genome: about 50% of affected individuals are assumed to represent de novo mutations (22). Neurofibromin is expressed in different cells, e.g. in glial and Schwann cells, as well as in melanocytes during certain stages of development (23). Neurofibromin is assigned to the GTPase-activating proteins (GAP) and exerts indirect effects on cell proliferation and differentiation (24). Loss of function or reduced expression of neurofibromin causes uncontrolled cell growth and leads to various diseases arising in this condition, including cancers associated with NF1. Malignancies in sporadic tumors not related to NF1 can also show NF1 mutations (25-27). However, the basic cellular changes as a prerequisite for tumor development in NF1 are still unknown. Despite the enormous impact of basic science on the fields of NF1 research and the establishment of screening tests to identify NF1 mutations, diagnosis for clinical purposes still is based on medical history, physical findings and selected imaging modalities (2, 4, 5). Diagnostic criteria for NF1 were suggested by the US National Institutes of Health following a Consensus Development Conference in 1988 (28). The criteria were updated by Gutmann et al. (5). At least 2 findings have to be applicable in an individual to establish a diagnosis of NF1 (Table I); OPG is a defining feature to establish NF1 diagnosis (5) (Table I). Nevertheless, the biological basis of OPG in NF1 is presently unknown.
Optic Pathway Glioma (OPG). The first publication on OPG associated with a disease that was later denominated NF1 was probably by Michel in 1873. Michel described a glial hyperplasia of the right optic nerve and chiasma opticum on necropsy in a boy who was also affected by elephantiasis (29). This description was published prior to Recklinghausen's seminal description of this disease (17). Later, Emanuel reported on optic nerve tumors in children associated with Recklinghausen‘s syndrome affecting both sides of the nerve that showed no malignant transformation (30). Davis presented 5 patients with Recklinghausen's disease who also had OPG. Davis described these tumors to be distinguishable from neurofibroma and classified them as gliomas (31). Further experience in the field of OPG diagnosis and therapy were communicated by Hoyt and Baghdassarian (32). This report was based on 36 patients with OPG. They declared surgical resection to be frequently ineffective to sufficiently reduce the pathologically increased orbital content and radiotherapy to be ineffective in local tumor control (32). Lewis et al. (33) analyzed the clinical findings of 217 NF1 patients investigated at Baylor College of Medicine, Houston, TX (USA) who had been subject to cranial computed tomography. About 15% of these patients presented with OPG. The prevalence of OPG in NF1 varies between 5 and ≥20%; most OPG appear to be asymptomatic (3, 7, 13, 33, 34, 35). Male and female individuals with NF1 are almost equally affected by this condition (36). The proportion of OPG relative to all brain tumors in childhood is estimated as 3-5% (37-39).
Sporadic OPG (non-NF1) vs. NF1-associated OPG. OPG are characteristic findings in NF1, but sporadic occurrence has been well-known for a long time (32). The severity of clinical findings and the progression of OPG differs depending on the genetic background of the affected individuals (40-43). Several publications point to the fact that syndrome-associated OPG shows a moderate progression compared to sporadically-occurring OPG (40, 41). However, this finding could be the result of analyzing biased data. Shamji et al. (41) add for consideration that in some studies, symptomatic non-NF1 OPG are assessed against asymptomatic NF1-associated OPG, with patients of the latter group being diagnosed during routine ophthalmological screening investigations (42, 43). Consensus exists that loss of visual acuity is the most striking ophthalmological finding in both groups (38, 40, 42, 44). Several authors registered a more frequent loss of visual acuity in sporadic OPG compared to syndromic OPG (40, 42, 45). However, a significant difference concerning this item was only rarely reported (38). Associated symptoms and findings were also reported with a putative effect on the natural history of PA (44, 46). In cases with sporadic OPG, further findings like hydrocephalus, nystagmus and strabismus predominated the clinical presentation. NF1-affected individuals were more frequently faced with proptosis of the eye and precocious puberty (44, 46). These differences in clinical features could give clues to the primary tumor localization and spread of OPG with reference to the genetic background of the affected individual. Rush et al. (47) and Wright et al. (48) came to the conclusion that children with NF1-associated OPG have a better prognosis than children with sporadic OPG concerning survival. This finding was in contradiction to another report (49). These studies were performed prior to the genetically-based differentiation of neurofibromatoses and later studies provided better substantiated statements. Singhal et al. (40) confirmed the more aggressive course of sporadic OPG. These authors investigated 34 patients (NF1 and non-NF1) and documented 7 cases of death in direct connection with OPGs. However, five fatalities occurred in the non-NF1 group (total number=17). The differences were not significantly different between both groups. The number of treated patients with OPG constitutes a further indicator of disease severity. Six patients of the non-NF1 group had to be subjected to therapy, in contrast to 3 NF1 patients (40). The number of treated non-NF1 patients was considerably higher in an earlier report (18/19 non-NF1 vs. 5/17 NF1) (44).
Different radiological localization and imaging characteristics were reported for sporadic vs. syndromic OPG (44). Kornreich et al. (50) revealed a predisposition of sporadic OPG to invade the chiasm. A cystic component was noted in 60% of space occupying lesions (50). A similar picture emerged from the study presented by Chateil et al. (42): all sporadic OPG affected the chiasm. NF1-associated OPG were localized predominantly in pre-chiasmatic sections of the optic nerve. Relatively small tumors were identified in the majority of cases in NF1 patients (38, 50). Astrup (43) also noted non-NF1 OPG to be diagnosed later, to have greater extension and show a more marked progression than OPG in NF1 affected individuals. However, these group differences give only a rough indication of the very broad spectrum of clinical findings in individual cases.
WHO classification and histological findings in OPG. OPG are predominantly pilocytic astrocytomas (PA) WHO grade 1 (2). However, advanced grade (3 or 4) gliomas of the optic nerve were occasionally reported in NF1 affected individuals (51, 52). PA are well-defined, slowly growing tumors that are diagnosed in NF1-affected individuals predominantly during the first two decades of life. Tumor progression cannot be estimated from grading but histological confirmation of diagnosis can be useful for the evaluation of treatment options in cases with rapid tumor growth, atypical localization or tumor growth during chemotherapy (51). The large variety of symptoms and findings caused by PA rely on the localization and extent of tumor, e.g. macrocephaly, endocrinopathy, and intracranial hypertension. The tumors are of whitish-brownish color, show elasticity on palpation, and microscopically, the alternate fibrous and microcystic regions constitute the biphasic growth pattern (53). Elongated, whirlwind-like arranged, uniform cells with low mitotic activity are characteristic for PA. Cytoplasmic eosinophilic granular bodies and worm-like Rosenthal fibers appear frequently in PA (53). Occasionally, a marked nuclear pleiomorphy and increased endothelial proliferation are diagnosed in PA that are not indicative for malignant transformation. In rare cases, calcifications can also be seen.
Abundant amounts of mucous inside the PA are possibly the cause of spontaneous tumor regression that is occasionally diagnosed (54). The first report of regression in a PA affecting the optic nerve was probably diagnosed using magnetic resonance imaging (55). Gottschalk et al. reported on 2 patients with chiasmatic gliomas close to the hypothalamus that showed spontaneous regression during the observation period of less than 24 months (56). MRI is the standard imaging technique to monitor PA (44). Parsa et al. (57) reported on this phenomenon in more detail based on a selected group of patients. They documented regression of tumor volume in 12 of 13 patients. However, regression was more frequently seen in sporadic (n=8) than in NF1-associated OPG (n=4). Parsa postulated that these findings are capable of generalization to derive and consequently categorize OPG as hamartoma (58-60). The frequency of spontaneous regression of OPG is unknown but it appears to be a rare phenomenon.
Radiological localization and imaging characteristics of OPG. The predominant localizations of PA are (53): optic nerve(s), chiasma opticum and hypothalamus, thalamus and basal ganglia, cerebral and cerebellar hemispheres, brainstem.
The topographical classification of Dodge et al. (61) can be used to describe the extension of OPG (grades I-III). Dodge I: tumor invasion is restricted to optic nerve, Dodge II: tumor invades the chiasm with or without invasion of the optic nerve(s), and Dodge III: tumor invades the optic tract with or without invasion of the hypothalamus.
MRI combined with the application of a contrast agent is the most preferred imaging modality to identify OPG (2, 5). Radiological characteristics of OPG are tubular thickenings on the one hand and the intraorbital winding route of the optic nerve with optional angulations (‘kinks’) on the other (2). OPG show enhancement following the application of contrast agents; however, differentiation between the optic nerve and the tumor is not possible when using MRI at 1.5 or 3.0 Tesla. OPG appear on MRI typically as iso- or hypointense structures on T1-weighted images, are signal-intense on T2-weighted sequences and show a homogenous contrast agent uptake (36, 39). Intra-canalicular and intracranial extension of OPG is easier to identify on MRI than on CT. Beam hardening artifacts can superimpose tumor and optic canal on CT. Furthermore, in CT, the OPG is depicted as a homogenous thickening without distinction between the nerve and nerve sheath. On the other hand, MRI may show these structures. Van Es et al. (62) pointed to the capacity of MRI in OPG diagnostics to allow the identification of a widened nerve sheath as a finding to discriminate an affected from a healthy nerve.
Visual acuity in OPG. The majority of OPG in NF1 is clinically inapparent. However, about 30-50% of NF1-patients with OPG show symptoms like reduced visual acuity, endocrinological abnormalities, disturbances of color vision, visual field defects, atrophies of the optic nerve or a pale papilla (35, 44, 63). Children with NF1 are at-risk of developing a symptomatic OPG predominantly until their 6th or 7th year of life (33, 34). However, in some cases, symptomatic OPG were diagnosed in adolescents and adults, indicating the variable growth patterns of these tumors (64). The topography of tumor appears to be the decisive factor for the ophthalmological course of the disease. Post-chiasmatic and OPG close to the hypothalamus cause significantly more frequent reduction of visual acuity (62%) than intraorbital or pre-chiasmatic lesions (32%) (63). Precocious puberty occurs in about 12% to 40% of children with chiasmatic OPG (35, 38, 65). It is assumed that lesions close to the hypothalamus affect the hypothalamo-hypophyseal-gonadal axis and thereby initiate puberty.
Several methods are applied for the medical assessment of visual functions, e.g. evaluation of vision, determination of the field of view, ophthalmoscopic investigation and visual evoked potentials (VEP). Determination of visual acuity is assessed as the most reliable method, but erroneous measurements can be obtained in children. The implementation of methods to obtain reliable and reproducible results poses the most prominent problem in this age group. Whether small children (<2 years) should be subjected to cranial MRI following the diagnosis of NF1 still remains unanswered to a large extent (66). However, consensus is established that ophthalmological tests to verify visual acuity in children come up against barriers. Wolsey et al. (67) revealed a sensitivity and specificity of 50% each for verification of visual acuity in children with and without OPG using VEP. VEP revealed higher specificity (73%) and sensitivity (86%) in this study on NF1-associated OPG to identify alterations of visual acuity rather than conventional visual acuity testing. According to the authors, sensitivity increased up to 93% if serial VEP testing was considered. Routinely applying MRI as a screening modality is controversial. An argument against the routine application of MRI is the well-established knowledge that radiological and clinical findings do not match sufficiently (68).
Up to the age of 7 to 10 years, ophthalmological investigations should be performed on an annual basis (66, 69). Visual acuity testing should ideally be adapted to the age of patients using different symbols, figures, numbers or letters (70). The usage of unified protocols for these tests was recommended, but is presently not commonly applied (70). Several factors should be taken into consideration: age-adapted test, investigation with corrected/non-corrected visual acuity, distance between patient and system and compliance of patient. The authors recommend the use of Teller acuity cards (TAC) or computer-based tests in order to use easily applicable tests that provide reproducible results. Listernick proposed an age-adapted test for patients with OPG (66) (Table II). Furthermore, other orbitotemporal findings can have an impact on visual acuity in NF1 (71).
Therapy of OPG. The main problem when deciding on therapy in OPG is the unpredictable biological tumor behavior of PA in this location (60). Stable volume or slow growth patterns are predominantly noted, but rapid growth spurts can be also found. The rationale for therapy can be based on the radiological progression of tumor volume, e.g. (orbital) tumor with (additional) extension to the hypothalamic region, or clinical findings, such as rapidly reduced vision, endocrinological alterations, impaired visual fields, and proptosis (72, 73). Indications for surgery may be related to different aspects, e.g. aesthetic reasons, tumor extension, compression of adjacent structures, or exophthalmos with loss of vision. Indeed, orbital tumors can become so extremely large and disfiguring that enucleation with consecutive epithetics will result in a more convenient aesthetic appearance. However, a prophylactic surgical resection of intraorbitally confined OPG is not justified as post-chiasmatic extension is rare (72, 73). On the other hand, enucleation of an almost blind eye can be necessary in cases when there is a danger of sympathetic ophthalmia following orbital trauma or surgery. However, the prevalence of sympathetic ophthalmia is extremely rare, calculated as 0.03 per 100,000 individuals per year (74), and therapeutic options are limited (75, 76).
Recommended visual acuity tests depending on age in early childhood (adapted from (66)).
Chiasmatic and hypothalamic glioma, with the risk of the development of hydrocephalus, e.g. compression of 3rd ventricle, can necessitate surgical intervention. However, chiasmatic surgery is associated with a high rate of complications; in particular, hypothalamic damage may occur. This complication can lead to complete loss of vision and pituitary incompetence (36, 77).
In some studies on OPG therapy, the application of radiotherapy was recommended in children older than 5 years. Total dosage up 54.4 Gray (Gy) in fractions of 1.6-1.8/d were used. Fractionation (1.6 Gy) and total dosage (40-45.2 Gy) were adjusted for therapy in small children (<5 years) and low-grade gliomas (LGG) (78). However, the application of radiotherapy has been controversially discussed in the past several years, because severe undesirable side-effects were noted in irradiated NF1 patients which can substantially impair the range of radiotherapy applications in NF1 patients. Complications like endocrinological, neuro-psychological and neurovascular diseases were also reported (79).
The development of malignant peripheral nerve sheath tumors in the former irradiation field is a severe complication in the course of treatment for OPG (80, 81). The five-year survival rate is poor in general (about 20%) and NF1 appears to be a predictor of poor prognosis in MPNST (81). However, only a few studies have addressed the impact of NF1 on MPNST prognosis, with particular reference to the orbital region. Sharif et al. (80) analyzed the side-effects of radiotherapy in NF1 patients with OPG. These authors disclosed a total of 12 secondary neoplasms in 9 of 18 (50%) NF1 patients irradiated for symptomatic optic pathway gliomas. Five of these 12 neoplasms were MPNST. Other brain tumors were 7 gliomas outside the optic tract (thalamus, hypothalamus, 3rd ventricle, temporal lobe). NF1 patients with OPG without radiotherapy were used as a control group. In this group, secondary neoplasms were observed markedly less frequently: a total of 8 (20%) of 40 patients with non-treated OPG developed a further 9 tumors (8 gliomas, 1 MPNST). Median time for the diagnosis of second tumors in this study was 14 years (range: 1-33 years). The relative risk of NF1 patients with irradiated OPG to develop second tumors was calculated as 3.04. Stereotactic fractionated radiotherapy is a further modality for the treatment of OPG in NF1 that is currently not very popular in the treatment of OPG (37, 66). In Germany, the Pediatric Oncology Society recommends radiotherapy as an option in children with LGG such as OPG, but explicitly points out the good prognosis of NF1-associated OPG in general and the undesirable side-effects of radiotherapy in this patient group (82). The severity of side-effects in long-term survivors of LGG therapy during childhood was confirmed by other groups (83). Besides the localization of brain tumors, multivariable models demonstrated radiation therapy to be a significant independent predictor of several impaired brain functions, such as hearing loss, growth hormone deficiency, abnormal thyroid function, and adrenocorticotropic hormone (ACTH) deficiency (83). NF1 patients and associated LGG are increasingly more regarded as a separate group and radiotherapy is not recommended anymore to treat this entity by several authors (66, 78). Indeed, a high level of therapy-related morbidity following irradiation of LGG is reported in grade 1 tumors (83).
Chemotherapy is an established therapeutic option to treat small children (<5 years) with NF1 and OPG as the first line adjuvant therapeutic option (68, 84). Carboplatin and vincristine are the preferred agents; a third chemotherapeutic agent is not recommended for OPG in NF1 (66). The undesired side-effect of carboplatin is the allergenic potency of this drug (7-78%) (78). Chemotherapy can avoid the deleterious effects of irradiation for OPG. However, severe side-effects of chemotherapy in OPG treatment have to be considered when deciding on therapy for OPG (83). Chemotherapy consists of two phases (induction phase, about 6 months; consolidation phase, about 12 months). Alternatively, other cytostatics can be used in cases with progression or when allergic reactions to drugs are noted (cisplatin, cyclophosphamide) (66).
A ‘wait and see’ strategy with regular ophthalmological controls is alternatively recommended, in particular in the vast majority of OPG that are not symptomatic (35).
Amblyopia.
Definition of amblyopia. Amblyopia is defined as an impaired visual acuity without clinical evidence for an organic cause or with evidence for an organic pathology that is insufficient to explain the amount of visual impairment (85). Haase (86) and Haase and Gräf (87) define amblyopia as a dysfunction of the sense of shape. Amblyopia develops despite the presence of physiological neural circuits as a result of stimulus deprivation or pathological binocular cooperation.
The leading symptom of amblyopia is decreased (best-corrected) visual acuity (Table III). For practical reasons, in adults and children older than 6 years of age, vision <0.8 (monocular eye test character presentation or single optotypes) is defined as subnormal and thus amblyopic (87). It is worth pointing out the term ‘relative amblyopia’, describing a combination of organic pathology and amblyopia in patients with reduced visual acuity. This term will be applied for discussion of the current findings.
Epidemiology of amblyopia. A previous ophthalmologic study in Hamburg on unilateral or bilateral strabismus in 830 children starting school revealed 52 children (6.3%) with this finding (88). About half of the cross-eyed children (44%) were also affected by amblyopia. The prevalence of amblyopia in children of this study group without strabismus (n=778) was about 2%. Risk factors for amblyopia are (87): strabismus, anisometropy, ametropy, astigmatism (amblyopia-caused refraction anomalies), family history of amblyopia, preterm birth, anomalies of lids (ptosis)/cloudiness of the refracting media, e.g. in the course of cataract (deprivation amblyopia), and nystagmus (nystagmus amblyopia).
The most frequent causes of amblyopia are strabismus (about 50%), refraction anomalies (about 20%) and a combination of these conditions (30%) (89).
Amblyopia associated with neurofibromatosis type 1. The prevalence of amblyopia in NF1 is not well documented. Ardagil et al. (90) revealed a higher frequency of amblyopia (10% vs. 1.3%) and anisometropia (16% vs. 2%) in NF1 affected patients compared to a control group. Strabismus as the initial ophthalmologic finding – in particular associated with OPG – is frequently reported in studies on NF1 (38, 40, 70-72, 91, 92). Estimations of the frequency of reduced vision in NF1 patients are more precise for individuals affected by plexiform neurofibroma in the orbital region, termed orbito-facial neurofibromatosis (71, 92). In these studies, more than 50% of NF1 patients with associated facial neurofibroma (plexiform neurofibroma) presented impaired visual acuity. Another study reported a reduced vision of the eye of the affected side in 85.7% of orbitotemporal neurofibromatosis (93). An earlier report revealed an association of plexiform neurofibroma with OPG, and glaucoma (35).
Physiological stages of developing visual acuity (based on (86)).
Focal abnormal signal intensity (FASI) in NF1. Specific cerebral lesions or changes of signal intensity identified on MRI were not selected to the canon of findings to establish diagnosis of NF1. However, some authors emphasized their diagnostic value for NF1 (94, 95), in particular in young patients. These lesions were formerly known as ‘unidentified bright objects’ (UBO) but now are preferentially termed ‘focal areas of (high) signal intensity’ or alternatively ‘focal abnormal signal intensities’ (FASI) and are seen as signal-intense foci on T2-weighted MRI (96). Differentiation to LGG may frequently be difficult to achieve, even with advanced imaging techniques. In some cases, surgical exploration and histological investigation is needed to come to a correct diagnosis (95, 97). FASI do not exert a mass effect, are not marked by contrast agents and are preferentially detected in the cerebellum, basal ganglia, and brainstem (98). Some authors support the hypothesis that FASI represent developmental disorders of the myelin sheaths (99, 100). FASI are a frequent finding in NF1-affected children undergoing cerebral MRI (43-93%). An absence of FASI in NF1 children during their first 2 years of life was reported to be a characteristic finding (95). FASI increase in size and number until the age of 12 years and are rarely seen in adults (101). FASI do not cause neurological symptoms, but a correlation with learning difficulties was noted if present (102). There is no known association of FASI and OPG in NF1.
Materials and Methods
The basis of this retrospective study was the medical files of the Neurofibromatosis outpatient center, Center of Rare Diseases, Eppendorf University Hospital. Files of 1,827 patients with an ascertained diagnosis of NF1 (5) were analyzed, including medical and surgical reports, ophthalmological assessments of visual acuity, and radiological reports of computed tomography (CT) or magnetic resonance imaging (MRI). Firstly, all MRI reports were evaluated for any report of an optic pathway glioma. In approximately every second patient, a cranial MRI had been performed (n=925). A further 50 patients had been subjected to cranial CT. In total, 1,948 MRIs were available for study. The other patients had no physical findings suspected for having abnormalities affecting the optic pathway. This study was approved by the Institutional Review Board of Eppendorf University Hospital as a prerequisite for a doctoral thesis (MAN).
It should be noted that many patients were referred for expert opinion by other physicians from all federal states of Germany and in some cases even from abroad. Many patients were regularly seen at the Neurofibromatosis Outpatient Center, but others only appeared for a single investigation. It is likely that patients with more complicated needs may have been referred for expert medical advice or had received selected information from informed lay groups. Therefore, we cannot exclude a biased sample. Furthermore, it cannot be excluded that in the patient group diagnosed as having no OPG as revealed on adequate cranial images, the follow-up would have disclosed some patients with newly developed OPG, although this event is considered rare (35). This limitation of the interpretation of results is particularly important for assessment of the prevalence of late-onset OPG. In addition, the proportion of very small children in the study population is likely underrepresented. Furthermore, due to the retrospective study design, important individual factors could not be considered, such as experience of the investigator in the treatment of NF1-affected children, standardization of investigation protocol during the entire examination period of the single patient, and the time between customizing new glasses and the ophthalmological investigation.
Radiological investigation. The radiological diagnosis of OPG was made following the recommendations of published references (34, 41):
Alterations of the signal intensity in iso-intense T1-weighted and hyper-intense T2-weighted images on MRI
Description of the optic nerve's course with typical kinking or elongation of the nerve cord
Assessment of the width (‘thickening’) and form (‘widening’) of the nerve
Uptake of contrast media (predominantly gadolinium DTPA (Magnevist®, Schering, Berlin, Germany))
Asymmetries between the optic nerves
Alterations of the radiological findings during the follow-up
In 695 of 925 patients, the Siemens 1.5 Tesla Magnetom® (63 SP/Symphony/Avanto, Siemens Healthcare, Erlangen, Germany) scanner was used. In these patients a standardized examination protocol was established by a team of radiologists experienced in the diagnosis of neurofibromatosis patients (CF). The 230 other MRIs were performed nationwide and collected in the NF1 outpatient database. The technical data of 625 patients investigated with the 1.5 Tesla Magnetom® were kept constant during the whole recruitment period: T1-weighted images were performed with echo times of 15-25 msec and acquisition and repetition <600 msec. Proton and T2-weighted sequences were performed at echo times of 80-120 ms and acquisition and repetition times >2 msec. Parameters 2-6 were also applied to CTs.
The radiological localization of OPG was registered according to the following scheme: unilateral and bilateral (with reference to optic nerve). ‘Laterality’ of OPG was not applied in tumors beyond the optic nerves but refers to topographic landmarks identifiable on MRI: chiasmatic, post-chiasmatic, pre-chiasmatic and chiasmatic, chiasmatic and post-chiasmatic, total optic pathway affected. The localizations were numbered 1 to 7 in anterior to posterior direction starting with unilateral optic nerve glioma as ‘1’. These distinctions allow OPG localization to be classified according to Dodge et al. (61).
FASI. All patients with OPG were evaluated for T2w hyperintensities of the brain (FASI). The radiological reports did not explicitly address this item in every single case and this lack of information was recorded as a “missing value” (including 8 patients with OPGs diagnosed on CT).
Clinical findings. Patients with OPG were assessed as symptomatic or asymptomatic according to the visual acuity and/or course of visual acuity, compared to the imaging findings. In nine patients with OPG, the finding ‘symptomatic’ or ‘asymptomatic’ could clearly be determined after analysis of reports, imaging findings and individual reports, but the initial ophthalmological findings at the time of onset of symptoms or therapy were not available. The following ophthalmological findings were evaluated as symptoms of OPG:
Reduced (best-corrected) visual acuity
Defects of pupillary reaction
Impaired or loss of vision in a visual field
Ophthalmological assessment of papilla (pallor, atrophy)
Visual evoked potential (VEP) in cases with inconclusive symptoms, suspected of being affected with OPG or radiological evidence of OPG
Endocrinological findings, such as pubertas precox, abnormal insulin like growth factor-1 (IGF-1-), follicle stimulating hormone (FSH-) or luteinizing hormone (LH-) values in patients with OPG involving the chiasma/hypothalamus suspected for being symptomatic for endocrinological abnormalities.
Visual field examinations included Goldmann perimetry and static perimetric investigations (Humphrey program 30-2). Investigation for amblyopia included inspection for refraction, strabismus, lid anomaly and/or clouding of refractory media. Refraction indices of ≥1.5 dpt. and astigmatism of ≥1.5 dpt. were evaluated as indicators of amblyopia.
Symptomatic patients were grouped according to treatment (T) or no-treatment (NT) for OPG. The term ‘treatment’ refers to any medical procedure intended to provide relief from the consequences of OPG in the individuals affected by this condition. Treatment modalities were further subtyped (chemotherapy (C), radiotherapy (R), surgery (S), and combinations of these measures).
The findings of patients undergoing therapy registered at the first neurological, endocrinological or ophthalmological investigations were summarized in order to allow comparison with the NT group. Visual acuity of NT patients is shown in box plots, presenting the values of the first and final investigation of the observation period. In the T-group, the assessments of visual acuity prior to therapy are used as the reference values.
Statistics. Correlation of findings with reference to symptomatic or non-symptomatic OPG and T2 weighted hyperintensities were calculated using the fourfold table and phi-coefficient according to Pearson. Data were collected in Excel® Version 10 (Microsoft Corp., Redmond WA, USA) data sheets, checked for plausibility, and further processed using SPSS® statistic program version 19 (International Business Machines, Armonk, NY, USA). Contingency tables were applied to calculate the strength of connectivity between initial and final findings. Significance niveau was calculated applying the χ-square test according to Pearson and the exact test according to Fisher. Level of significance was defined α=0.05. p-Values <0.05 were defined as statistically significant.
Results
Of the 1,948 magnetic resonance images (MRI) of 925 patients, OPG were detected in 126 patients. A further 8 OPG were detected on computed tomograms (CT) of 50 patients. The total number of patients evaluated for OPG by cross-sectional imaging methods was 975. OPG were diagnosed in 134 patients (13.7%). Frequencies of OPG with respect to imaging technique differed only slightly (MRI: 13.5%, CT: 16%). The female/male ratio of NF1 patients with OPG was 70:64. In 799 patients, neither MRI reports nor medical files were indicative for OPG (Figures 1 and 2).
The total group (n=134) consisted of 57 symptomatic patients, 52 of them with evaluable data (missing: 5). Thirty symptomatic patients were treated and 22 symptomatic patients were not treated. Sixty-nine patients were not symptomatic for OPG. In eight non-symptomatic patients, data were judged to be incomplete for detailed analysis. One further non-symptomatic patient was categorized as being ‘treated’ due to chemotherapy for low grade glioma of the medulla oblongata. Therefore, the groups of ‘non-symptomatic’ and ‘non-treated’ patients show large overlap, but do not meet exactly to each other. On the other hand, the terms ‘treated’ and ‘symptomatic’ have to be clearly distinguished.
In 57% (n=77) of our OPG patient group, vision was not impaired. About 42.5% (n=57) of the total OPG group showed an unfavorable course of disease (i.e. were ‘symptomatic’), 29 of them were female (41% of the female group). The female to male ratio of symptomatic patients showed no preference for gender. The data are shown in Figures 3 and 4.
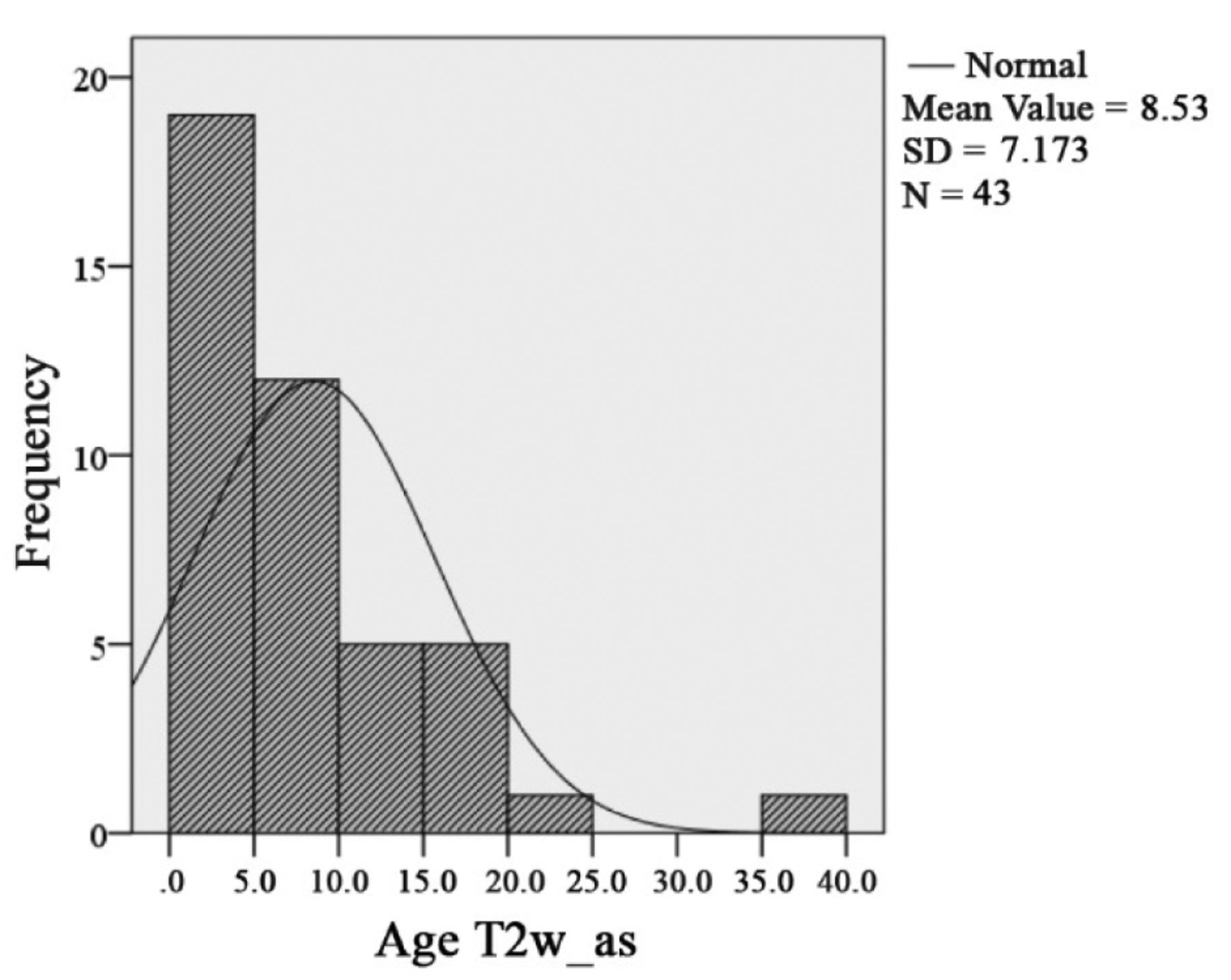
Mean age at the time of diagnosis of asymptomatic OPG was 11.6 years (SD=11.2, median=7.4; n=76, missing value (mv)=1). Sixteen of these 77 patients with an MRI-based diagnosis were >18 years old. The mean age of OPG diagnosis was 7.3 years (SD=4.5), considering patients younger than 18 years only (Figure 5).
The mean age of patients with symptomatic OPG at the time of diagnosis was 7.6 years (SD=6.92, median=6.1). This group included two individuals who developed symptomatic OPG aged 25 and 44 years. The arithmetic mean age is 6.4 years for initial diagnosis of symptomatic OPG in patients younger than 18 years (n=46) (Figure 6).
In addition to reduced vision, the predominant initial ophthalmological finding is strabismus (Figure 7). Findings constituting the diagnosis of amblyopia in this study group are further specified (see below). The distribution of (relative) amblyopia between OPG patients with or without treatment was statistically insignificant (p=0.548 (Pearson), 0.721 (Fisher)).
Table IV summarizes the comparison of initial ophthalmological, endocrinological and fundoscopic findings in treatment (T) and no-treatment (NT) symptomatic OPG patients (n=48). Interestingly, 11 of 48 patients (23%) presented with protrusion of the globe at the time of initial ophthalmological investigation. One patient in the NT group presented with unilateral protrusion at the age of 6.5 years and showed stable visual acuity during an observation period of 75 months (vision: 1.0). However, the item ‘protrusion of eye ball’ did not differ significantly between the groups (Table IV).
Seven patients (15%) showed endocrinological abnormalities. Deviations from the expected timing of sexual development accounted for 5 patients (pubertas praecox 4, pubertas tarda 1), while in a further 2 patients, hormone synthesis was affected (IGF-1R, ACTH, LH, FSH). Two patients experienced diabetes insipidus after surgical intervention. One patient developed pituitary insufficiency after radiotherapy for OPG.
Ophthalmoscopic findings revealed no difference between the T- and NT-groups. However, pallor of the papilla was registered in 63 of the 88 investigated eyes of this subgroup (72%) (Table IV).
Table V summarizes the findings that are relevant for establishing a diagnosis of amblyopia (n=10). Strabismus is the most prominent finding associated with amblyopia (n=7); combinations of several causes constitutive for amblyopia were diagnosed in 3. In 5 patients, ptosis was diagnosed, which was only slightly established in one, but it was relevant for relative amblyopia in 4 patients. In patients NT5 and R6, the ptosis was noted prior to OPG diagnosis.
The visual acuity of NT patients is shown in Figure 8. The visual acuity is separately listed for the better and worse seeing eyes in patients with symptomatic OPG. The interquartile distance for the better seeing eyes was initially 0.5 to 1.0 and at the end of the observation period was 0.65 to 1.0. The parameters for the worse seeing eyes were initially 0.15 to 0.7 and at the end of the observation period 0.15 to 0.7 (median=0.4). The median of the better seeing eyes improved from 0.75 to 1.0. These findings are also separately listed according to age group (Figures 9 and 10).
The ophthalmological findings in the treatment (T) group varied considerably and did not show statistically significant differences of treatment effects on vision. Therefore, the findings of these patients are listed in detail in Table VI.
Localization of OPG in symptomatic and asymptomatic patients (MRI). The exact localization of OPG was determined in 128 patients (mv=6). In 14 (27%) patients with unfavorable follow-up, the entire optic pathway was affected. In 2 (2.6%) of the patients with favorable course, the glioma also affected the complete pathway. These differences were highly significant (p<0.0001, chi-square). In 27 patients (35.5%) with asymptomatic OPG and 9 (17.3%) of symptomatic OPG, the optic nerve was unilaterally affected (p=0.024). Bilateral OPGs were diagnosed in symptomatic and asymptomatic OPG with frequencies of 9.6% and 22.4%, respectively. About 45% of OPG involved the pre-chiasmatic parts of the optic pathway, including both symptomatic and asymptomatic patients. The proportion of chiasmatically localized OPG was the same in both groups, accounting for seven individuals each. Comparison of all other locations proved no significant differences of topography with respect to a symptomatic or asymptomatic OPG. Strictly post-chiasmatically localized OPG were not noted in this study (Tables VII and VIII, Figures 11 and 12).

Frequency of optic pathway glioma in a sample of 925 NF1 patients with cranial magnetic resonance images.

Frequency of symptomatic (sympt) and asymptomatic (asympt) optic pathway glioma (OPG) revealed on magnetic resonance images and computed tomograms (n=134).
Comparison of initial ophthalmological, endocrinological and fundoscopic symptoms and findings between both groups of patients affected with optic pathway glioma
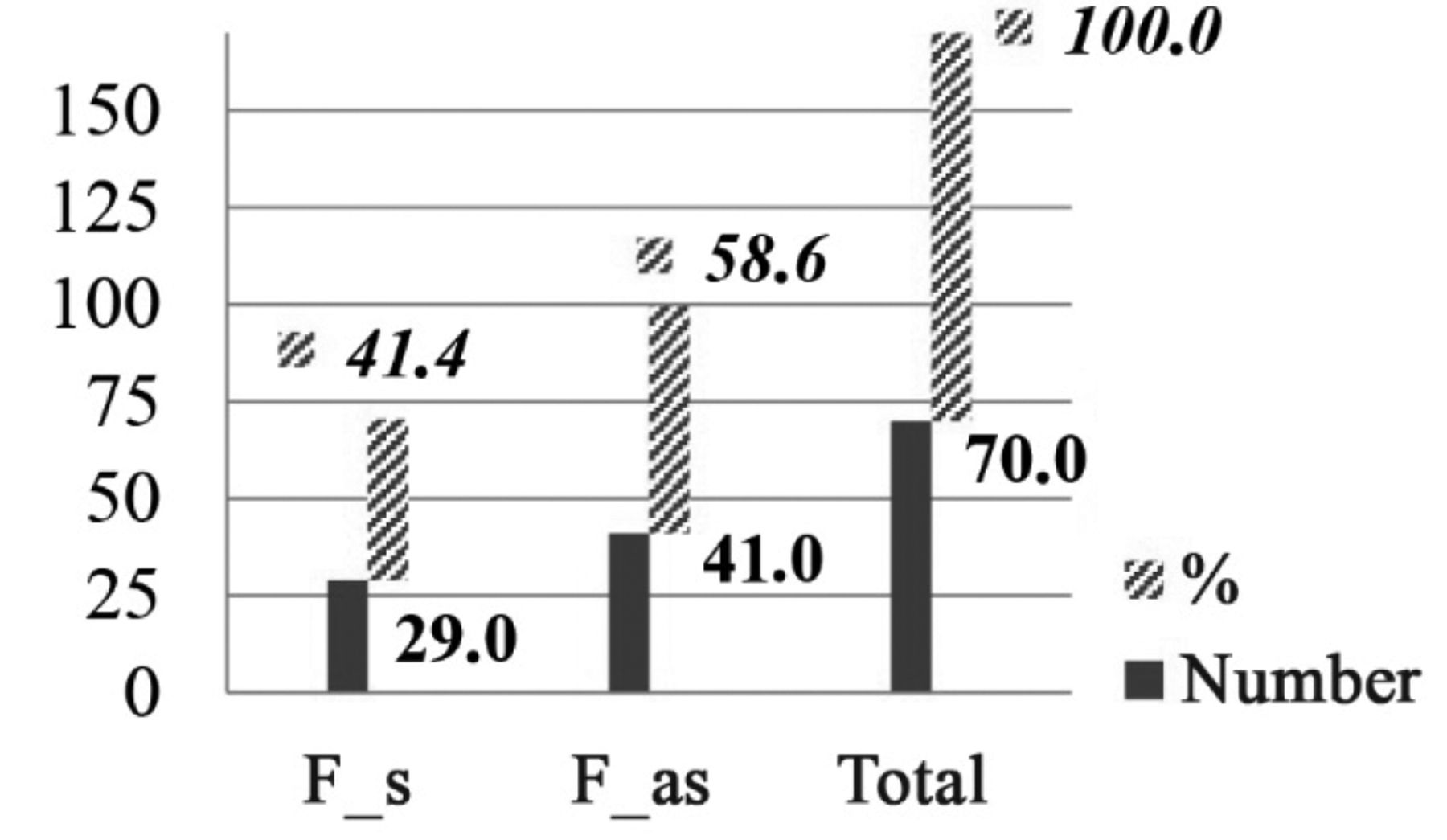
Sex ratio in relationship to symptomatic (F_s) and asymptomatic (F_as) optic pathway glioma in absolute and relative number (%) in females (F).

Sex ratio in relationship to symptomatic (M_s) and asymptomatic (M_as) optic pathway glioma in absolute and relative number (%) in males (M).
Surgery. Partial resection of the optic nerve was the most frequent surgical procedure (n=7). Complete resection was performed in 3 patients. In one patient, extirpation of the optic nerve at the chiasm was initially chosen, but progressive exophthalmos required enculeatio bulbi. Patient S6 died 27 months after initial ophthalmological presentation following resection of hypothalamic glioma and consecutive pituitary insufficiency. Six patients of the surgically treated group showed initially unilateral exophthalmos. Five patients lost their vision on the side of surgically treated OPG.
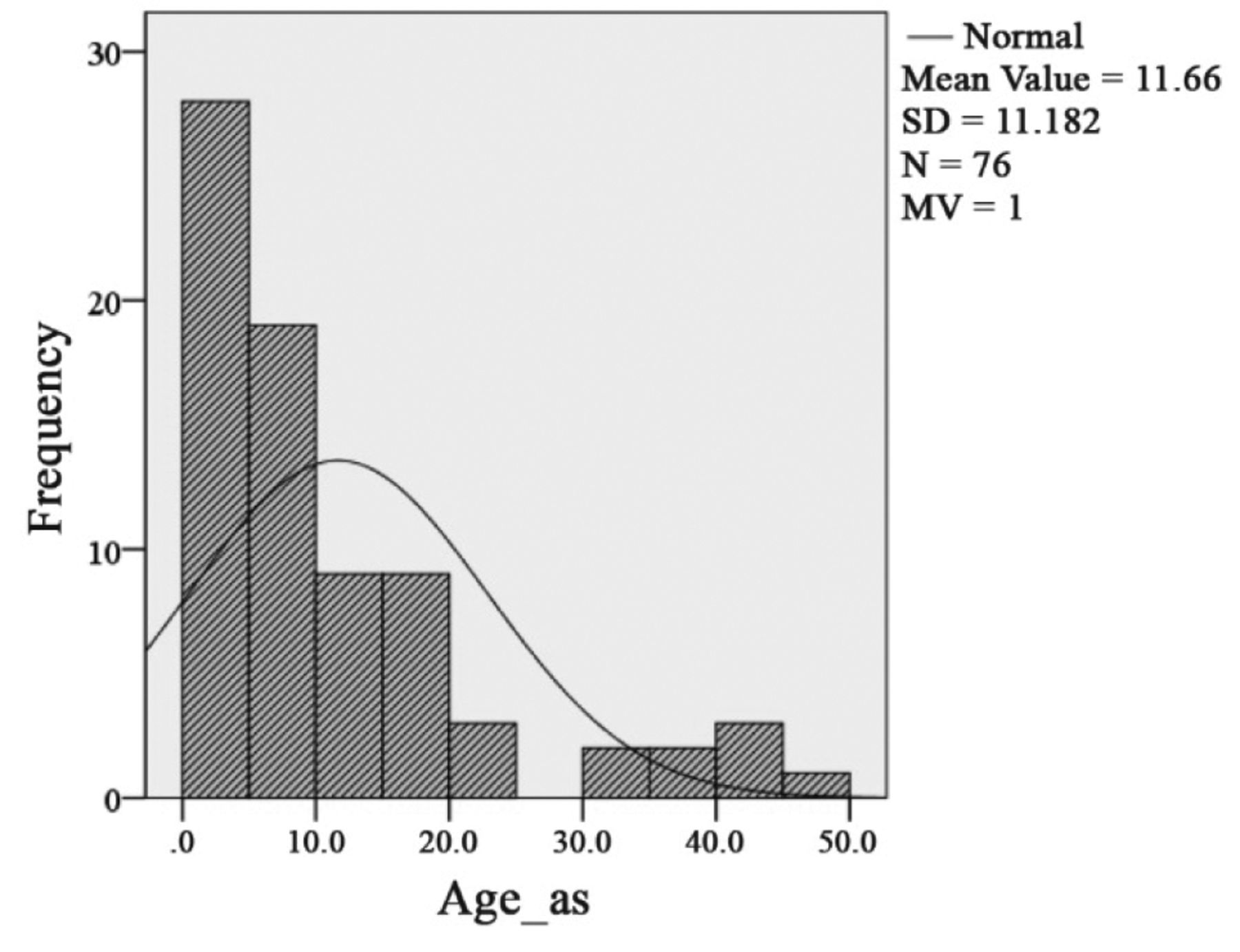
Histogram of age distribution of asymptomatic (Age_as) NF1 patients with optic pathway glioma in relation to a Gaussian distribution.

Histogram of age distribution of symptomatic (Age_s) NF1 patients with optic pathway glioma in relation to a Gaussian distribution.

Initial clinical findings of symptomatic optic pathway glioma patients in absolute and relative (%) numbers (n=48; missing values=9) (multiple designation possible), ED=endocrine deviation.
Chemotherapy. One patient was surgically explored for nerve sheath myxoma prior to diagnosis of OPG. In this case, tumor progression and loss of vision justified the use of chemotherapeutic agents for OPG treatment at the age of 6 years. In 2 patients, allergies to chemotherapy were noted (vincristine, carboplatin). Patient C6 developed further intracerebral astrocytomas in addition to the chemotherapeutically treated glioma of the optic nerves and chiasm.
Radiotherapy. History of external radiotherapy was noted in a subgroup of patients, but detailed specification for radiation protocol were not available. Biopsies were performed in 4 irradiated patients (Patient No. R3, R4, R5 and R6) prior to therapy. Diagnosis was astrocytoma WHO grade 1 in three patients, and grade 1-2 in one.
Focal abnormal signal intensity (FASI). FASI were noted in 71 of 134 OPG patients (53%). Out of 57 symptomatic OPG patients, 28 (49%) showed one or more FASI. Mean age of diagnosis of FASI was 6.94 years in symptomatic patients (SD 4.15). One or more FASI were diagnosed in 43 (56%) of asymptomatic OPG patients. The mean age of FASI diagnosis was 8.5 years (SD=7.17) in asymptomatic OPG patients. Differences in the distribution of T2W hyperintensities between symptomatic and asymptomatic OPG patients was insignificant (p=0.942). There is no correlation of FASI and OPG in NF1 (Figures 13, 14, 15 and 16). Findings of OPG studies in NF1 patients are summarized in Table IX.
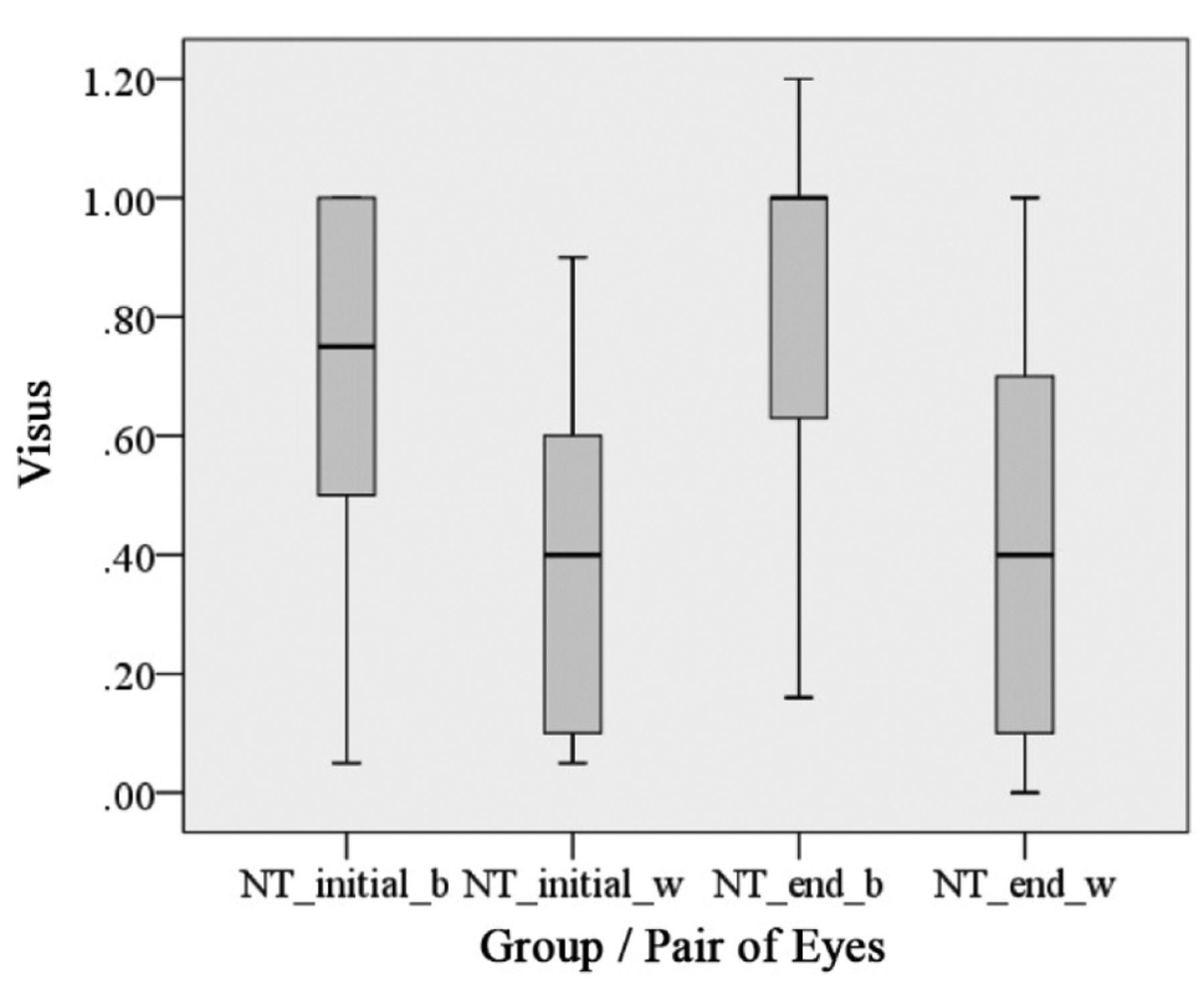
Descriptive illustration of visual acuity of NF1 patients with optic pathway glioma without therapy (NT group) in the beginning (_initial) and at the end (_end) of the observation period (6 months – 240 months). Visual acuity measurements are separately displayed for the better (_b) seeing eye and also for the eye with worse visual acuity (_w) (n=20, 40 eyes).
Synopsis of findings relevant to diagnose ‘amblyopia’ that could have an impact on visual acuity in neurofibromatosis type 1 patients with OPG.

Visual acuity of both better and worse seeing eye in relation to age group in the beginning of the observation period in patients with symptomatic OPG who were not treated (NT group) (6 months - 240 months; n=20, 40 eyes; *extreme value).

Visual acuity of both better and worse seeing eye in relation to age group at the end of the observation period in patients with symptomatic OPG who were not treated (NT group) (6 months - 240 months; n=20, 40 eyes; °measured value outlier).
Discussion
This study shows the high variability of the clinical course of OPG in patients with NF1 and the wide wingspan of therapies in symptomatic patients. A thorough documentation of the clinical findings and the collection of data from large study groups is necessary to define this phenotype in the disease.
Clinical course of Neurofibromatosis type 1-patients treated by chemotherapy (C), radiotherapy R or surgery (S) with pre- and post-therapeutic visual acuity.
Prevalence. OPG is a defining feature when diagnosing NF1 (5). Medical consultation of patients and relatives is difficult in affected individuals concerning the prognosis and therapy of OPG. This study analyzed a large number of medical reports of NF1-affected individuals and detailed diagnostic and therapeutic measures in 134 affected individuals. The prevalence of OPG in NF1 is 13.7% based on MRI-proven orbital diagnosis in 925 affected individuals and this proportion of affected patients in the total group is in accordance with current estimations of OPG prevalence in the context of NF1, in particular compared to the studies of White et al. (14%) (48), Lewis et al. (15%) (33) Listernick et al. (15%, 19%) (34, 35) and Segal et al. (13%) (103). A brief review of some previous studies on OPG in NF1 is provided in Table IX. Further publications calculated a prevalence of OPG in NF1 in the range from 5% to more than 20% (13, 40).
Radiological localization of symptomatic and asymptomatic optic pathway gliomas with respect to Dodge classification.
Huson et al. performed a population-based study on the prevalence of NF1 in a defined territory of the United Kingdom (104, 105). They found 2 of 135 NF1 patients affected with OPG. However, this study did not include asymptomatic patients. Lewis et al. (33) identified 33 OPGs in 217 patients with NF1, allowing the estimation of OPG prevalence in this entity by 15%. This study was the first in a series of well-defined studies applying strict diagnostic criteria for both the syndrome and the diagnosis of OPG. The assumption of a prevalence of 15% in NF1 was substantiated by consecutive studies, using both CT and MRI (34, 35). A review of OPG in NF1 recently claimed a prevalence of OPG in NF1 to be about 20% (66). Indeed, a study of Blazo et al. (106) based on meticulous ophthalmological findings and MRI registered OPG in about 20% of their NF1 patients (11 of 54 patients: the percentage of OPG in NF1 would have been even higher if only patients had been included that had been subjected to complete cranial imaging (106)). About 50% of OPGs in NF1 are detected in patients <10 years of age (48, 103).
Tumor biology. Tumor biology in NF1-associated OPG and sporadic OPG was repeatedly compared (35). The clinical findings at the time of initial diagnosis are crucial for estimation of tumor biology (83). Some studies compare symptomatic sporadic cases with asymptomatic OPG in the NF1 group (41).
The study by Kornreich et al. (50) showed a predisposition of sporadic OPG to infiltrate the optic chiasm; cystic areas were diagnosed in 60% of this group. A similar clinical picture arises from the results published by Chateil et al. (42) with tumorous invasion of the chiasma in all sporadic OPG. NF1-associated OPG were predominantly restricted to pre-chiasmatic parts of the optic nerve. Small tumors were primarily diagnosed in NF1 patients (38, 43, 50).
OPG and gender. In early reports on OPG in NF1, the male/female ratio was 1:2 (34). A further study revealed a female/male ratio of 1.4 (106), similar to the findings in the study published by Kluwe et al. (107). A more recent study showed a minor predominance of males affected by OPG in both sporadic and syndromic cases (1.15:1). However, most studies assume a balanced distribution of OPG between males and females (33, 34). The present study confirms the almost balanced prevalence of OPG in males and females (1.1:1), supporting earlier estimations by our center that OPG manifestation is independent of gender (91).
Age at time of diagnosis as a prognostic indicator in children and adults. Sporadic and NF1-associated OPG are predominantly diagnosed in childhood. Listernick et al. (34, 35, 44) noticed the maximum risk time to develop symptomatic OPG in children with NF1 younger than 6 years of age. The mean age of OPG diagnosis in children was 4.2 ys. The median age for these patients was 1.9 ys. In contrast to these findings, asymptomatic OPG were diagnosed about 3 years later in the children's lives (mean=5.3 years, range=0.7-20 years) (35).
A similar study from Germany (91) on NF1 patients reported differences between the mean age at the time of diagnosis depending on the clinical course (poor prognosis: 3.2 years, good prognosis: 5.8 years). Earlier publications stated higher ages at the time of OPG diagnosis (10-11 years) (47, 48). These differences are likely to address the impact of recruiting conditions and investigation methods on the accuracy of diagnosis.
Survey of all clinical and radiological findings of neurofibromatosis type 1 patients with symptomatic OPG at the time of first (ophthalmological) diagnosis (missing values: n=9).
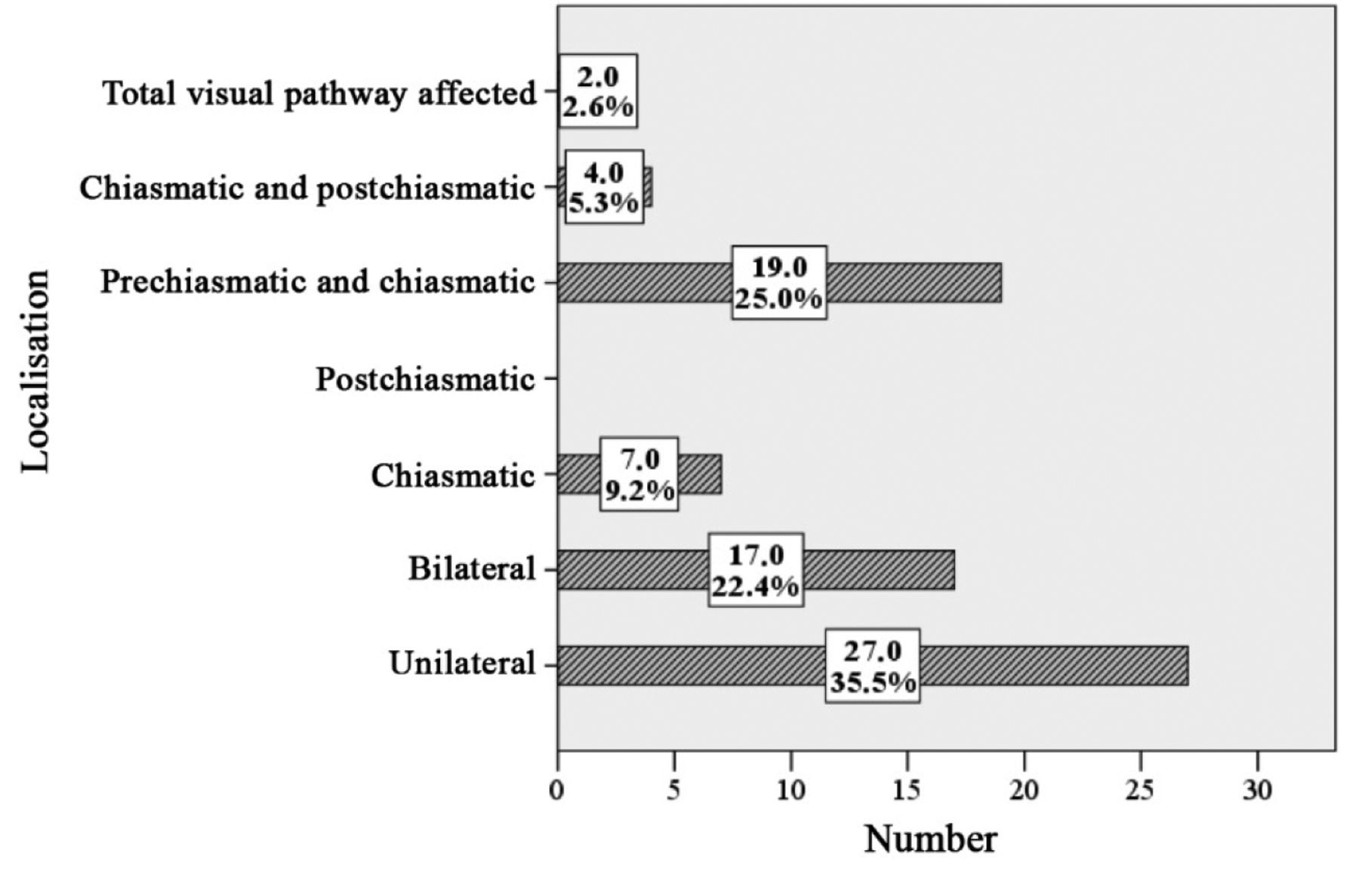
Radiological localization of asymptomatic optic pathway glioma (absolute and relative numbers) (n=76, missing value=1).

Radiological localization of symptomatic optic pathway glioma (absolute and relative numbers) (n=52, missing value=5).

Number of patients with symptomatic (T2w_symptomatic) optic pathway glioma and FASI detected on MRI.

Number of patients with asymptomatic (T2w_asymptomatic) optic pathway glioma and FASI detected on MRI.
Review of selected references presenting ophthalmological and endocrinological findings in non-NF1 and NF1 associated OPG with reference to statistically significant differences between both groups (p).
In this study, the mean age at the time of diagnosis was 6.4 years in children and adolescents with symptomatic OPG. Our data match exactly with reports from the literature. Integration of all patients (>18 years) to calculate this item increases the mean age at diagnosis up to 7.6 years of age in symptomatic NF1-associated OPG.

Histogram of age distribution of NF1 patients with FASI and symptomatic (T2w_s) optic pathway glioma.
Patients with asymptomatic OPG were diagnosed at an older age (11.6 years). This finding is related to the high proportion of asymptomatic OPG diagnosed in adults on MRI (16/77, 20.7%). Thiagalingam et al. (79) demonstrated asymptomatic OPG to be diagnosed about 5 years later than symptomatic OPG. Their finding is in accordance with the results of the present study. On the other hand, Segal et al. (103) noted in their study group an almost identical mean age at the time of primary OPG diagnosis, independent from the clinical course of the tumor. Whereas the majority of NF1-associated OPG occur during childhood, there is a substantial risk of patients with NF1 developing a symptomatic tumor in later periods of life (64). Three out of 43 NF1 patients with OPG developed their tumor in adolescence and experienced a substantial impairment of visual acuity despite prior ophthalmological investigations that had proved an asymptomatic course (63). A second study reported the case of symptomatic OPG in an 18 year-old patient (91). The present study adds two patients with symptomatic OPG becoming for the first time symptomatic in adulthood (Table VI).
Data from the literature and the present results demonstrate ophthalmological abnormalities or MRI findings leading to a diagnosis of OPG is usually established on average in the 7th year of life. The proportion of 16 newly diagnosed adult NF1 patients with asymptomatic gliomas support the hypothesis that some OPG remain unsuspicious for long periods of time. In addition, spontaneous regression of OPG was noted in one case of this study. Further reports on OPG in adults underline the biological variability of PA. OPG in NF1 is a finding that can become symptomatic far beyond childhood, at least in some cases. Therefore, an ophthalmological screening program in adult NF1 patients with asymptomatic OPG should be considered.
Histogram of age distribution of NF1 patients with FASI and asymptomatic (T2w_as) optic pathway glioma.
The age distribution of symptomatic OPG covers a wide range (1¾ years up to 44 years). This finding reinforces the specific challenges of age-adapted ophthalmological OPG diagnostics.
Diagnostic methods and screening for OPG. There are several methods applicable to study vision. The focus of studies is on visual acuity tests, field of vision tests, and visual evoked potentials. The most important test is determination of visual acuity. This parameter is used in many follow-up control studies. The comparability of results would enormously benefit from standardized procedures and their interpretation (70). Impaired field of vision is frequently correlated with reduced visual acuity in OPG of NF1 patients (72).
There are different opinions on whether ophthalmologic screening techniques are necessary in NF1 children with an unsuspicious course. Guidelines of the United Kingdom propose screening in asymptomatic children with NF1 and OPG until the 7th year of life (66). Other research groups do not limit the risk period to early childhood and propose a long-term follow-up control (10-25 years) (69, 72).
MRI is a generally recognized examination technique for screening of OPG (106). However, Ferner et al. (66) give account to the low rate of initially asymptomatic OPG becoming symptomatic and requiring chemotherapy during a yearly performed cranial MRI (106). Furthermore, they indicate that two children had received obviously unnecessary chemotherapy as a consequence of radiological imaging of OPG despite a stable ophthalmological investigation.
On suspicion of OPG, MRI is the preferred imaging technique to visualize the optic pathway. However, systematic MRI screening for OPG in NF1 children is a controversial subject: the majority of OPG are asymptomatic and do not require therapy and the natural course of a diagnosed OPG cannot be derived from MRI findings. Furthermore, the appropriate technical and economic means are high, and the risks of anesthesia for small children in OPG diagnosis have to be considered (72, 108). Therefore, systematic screening by means of MRI should correlate to clear ophthalmological findings and symptoms. Unfortunately, some ophthalmological findings in patients with OPG have a dubious character but cannot be used as a safe diagnostic parameter for determining a therapy. The predominant findings and symptoms (loss of vision, fundoscopic alterations, protrusio bulbi, strabismus) were detailed in the present study. The high percentage of pathologic fundoscopic findings (72%) on initial ophthalmologic investigation could provide an important predictor for symptomatic courses. Campagna et al. came to a similar conclusion (109). These authors considered a marked pallor of the papilla an adverse factor in the further course of OPG. Therefore, in cases of suspicion for OPG, a thorough ophthalmological investigation, based on standardized procedures, should be carried out, preferentially in specialized centers (2, 70, 72). Our results confirm previous estimates that stable vision over time in NF1-associated OPG cannot be predicted by any diagnostic aid despite early diagnosis, a long period of unchanged shape and extension of OPG on MRI, and constant visual acuity. The difficulty in assessing the underlying variable tumor biology with the aid of currently available diagnostic measures is an important reason for the reluctant strategies in the treatment of OPG in NF1 (68, 109). In summary, in selected cases, detailed screening with MRI may be useful, e.g. in the case of suspicion for OPG and unreliable results of ophthalmologic investigations. This could be the case in small children (72).
In some retrospective studies, VEP was proposed as a sensitive screening tool to diagnose OPG (sensitivity up to 93%) (67, 110). A drawback of the latter method is that small alterations of the amplitudes have a great influence on the evaluation of the results and can cause misinterpretations. Furthermore, poor compliance of patients influences the recordings. Some authors criticize the frequently applied retrospective study design and the implementation of these tests in cases with knowledge of already established diagnosis of OPG. The clinical circumstances should be considered when discussing the high sensitivity rate of VEP (72).
Findings of NF1 patients with OPG excepting impaired vision. The general assessment of OPG states that asymptomatic courses constitute the rule. However, about 30-50% of NF1 patients with OPG will develop tumor-associated symptoms (impaired vision, pubertas praecox, etc.) (35, 44, 63, 103). The results of present study support the thesis that only the simple majority of OPG will have an inapparent course (66). Indeed, the proportion of 43% patients with symptomatic OPG is high. The two predominant clinical findings at the time of OPG diagnosis were strabismus (n=12) and exophthalmos (n=11).
Strabismus. Strabismus is frequently reported an initial finding in OPG diagnosis (111). Ophthalmological diagnosis should consider both glioma and orbital plexiform neurofibroma as a cause for strabismus (71, 93). Causality of strabismus should be thoroughly evaluated in NF1 patients, in particular in the case of associated reduced vision. In our patients with amblyopia (n=10), 4 patients were diagnosed with relative amblyopia. Amblyopia is frequently diagnosed in early childhood. In this study, amblyopia was diagnosed in 10/48 patients (20.8%). Strabismus (7/10) is the prominent factor associated with amblyopia in this group of patients. Further factors are ptosis (n=4) and anisometropia (n=2).
Ardagil et al. (90) described a higher prevalence of anisometropia and amblyopia in NF1 patients compared to non-NF1 patients. Prevalence of amblyopia was rated as 10% in NF1, and 1.3% in the control group. A limitation of the study was the small number of patients included for investigation. Further studies are required to support the hypothesis of increased danger of amblyopia in NF1. The impact of amblyopia on vision was also stressed in the paper of Oystreck et al. (92) on (plexiform) neurofibromas of the orbitofacial region. In this report, one or more types of amblyopia were registered as a cause for reduced visual acuity in 39/55 NF1 patients. The impact of additionally diagnosed OPG (9/55 patients) proved to be irrelevant for the impaired visual acuity in patients with orbitotemporal plexiform neurofibroma in that study (92). The impact of attention deficits in NF1 affected children on the test results is not considered in these studies.
As a consequence of these results, the appropriateness of including NF1 patients with OPG in a single study group should be discussed. Indeed, numerous disease-associated findings can have an impact on the vision of an eye that is affected by OPG. In order to clarify the reasons for the wide variety of findings and symptoms in OPG of NF1, it is at least worth thinking about prospective studies on NF1-associated OPG that consider other findings in detail that may interfere with vision (glaucoma, strabismus, ptosis, etc.).
Exophthalmos. Intraorbital situated OPG cause frequently an exophthalmos. Studies investigating this finding allow the assumption that about 30% of symptomatic OPG cause exophthalmos (35, 112). Varan et al. (111) determined in their study on OPG (NF1 and non-NF1) exophthalmos in about 10%.
Skeletal anomalies of the orbital region can be associated with exophthalmos. Orbital plexiform neurofibroma (PNF) can also cause exophthalmos in an orbit of normal size (113). Indeed, orbital dysplasia can develop as a solitary osseous dysplasia or in close association with the development of soft tissue tumors of the orbit and periorbital region (114). Orbital and eye lid PNF frequently involve adjacent structures and PNF occasionally infiltrates and destroys these structures, e.g. the optic nerve or the eyeball (93). Protrusion of the temporal lobe into the orbit following sphenoid wing dysplasia can cause pulsating exophthalmos (115). Orbital PNF as the cause of exophthalmos was excluded in the present study on cross-sectional images. However, some patients had eyelid PNF that caused ptosis (Table VIII). Sphenoid wing dysplasia also was an infrequent finding in this study group as was anterior orbital extension of eyelid PNF (Table VIII).
The results of the present study lie in a range between the previously mentioned percentages. Exophthalmos was diagnosed in 11 of 48 (23%) patients with symptomatic OPG. Exophthalmos and OPG are frequently indications of extensive tumor and/or progression of OPG. Functional and aesthetic impairment are strongly associated with this finding. The comparison between treated and non-treated OPG patients revealed no significant difference concerning the distribution of the finding ‘exophthalmos’.
Ptosis. The frequency of congenital ptosis in NF1 varies between 3.7% and 9% (116, 117). Ptosis can occur independently of other orbital pathologies in NF1. The medical reports of this study were not suitable to clarify possible congenital ptosis as a cause of visual impairment in OPG affected individuals. The finding ‘ptosis’ was recorded rarely in the reports of the present study patients.
Endocrinological disorders. Extension of OPG to the optic chiasma can cause endocrine system alterations (pubertas praecox, diabetes insipidus). Deviation from normal pubertal growth were noted in 5/48 patients, and in a further two patients alterations of hormone production were diagnosed (LH, FSH, thyroid stimulating hormone (TSH), ACTH, IGF-1). Three further endocrine system disorders were recorded during two surgical interventions) (diabetes insipidus) and consecutive to external irradiation (pituitary insufficiency). A large NF1 study recorded pubertas praecox in 7 of 219 (3%) children. Early puberty can be an important criterion to diagnose OPG of the chiasma in patients with NF1 (64, 65). Similar results were provided by a study from Italy that reported pubertas precox in 2.4% of patients with NF1 (118). However, higher frequencies were also recorded in children with supra-sellar gliomas (12% to 40% (38)). A retrospective study on OPG affected patients reported in 32 of 54 individuals the presence of pathological ophthalmological findings at the time of initial diagnosis of OPG. The predominant finding was reduced vision (72%) and exophthalmos (31%). Protrusion of the eye was frequently diagnosed in small children (<6 years), pubertas praecox was exclusively diagnosed in NF1 affected individuals > 6 years of age (79).
Radiological localization of OPG as a predictor of prognosis. There are different views on the predictive value of topography on the course of OPG. Some authors stress the poor course of OPG in tumors affecting the chiasmatic and post-chiasmatic optic pathway (66). On the other hand, Schröder et al. (91) pointed to the fact that chiasmatic OPG can show a favorable course and topography cannot be used as a reliable predictor for prognosis. Closer inspection of the topographic distribution in symptomatic and asymptomatic OPG in NF1 reveals significant differences:
Intraorbital glioma. Bilateral OPG are a typical finding in NF1 (119). The general assumption was confirmed to diagnose OPG of NF1 patients in the optic nerve(s) more frequently than the chiasm (44, 46). In our study (n=128, missing values=6) bilateral optic pathway gliomas accounted for about 17%. An impressive difference was noted for the association of bilateral OPG and the presence of visual symptoms. Seventeen of 76 patients (ca. 22.3%) with asymptomatic OPG were affected by bilateral OPG but only five of 52 (ca. 9.6%) individuals constituting symptomatic OPG group. In about double the frequency (27/76 OPG_as vs. 9/52 OPG_s, i.e. 35.5% vs. 17.3%), unilateral OPG were diagnosed in asymptomatic individuals with NF1.
In about 45.3% of patients with symptomatic or asymptomatic OPG, a space occupying lesion (either unilateral or bilateral) was diagnosed that did not involve the optic chiasma. Forty-four of 128 OPG showed an asymptomatic tumor (OPG) situated along the intraorbital parts of the visual pathway (Dodge classification I). On the other hand, 14 of symptomatic patients of this total group showed intraorbitally localized tumors, either unilateral or bilateral. Published findings of Segal et al. (103) present a similar distribution of Dodge-I classified OPG in the whole study group of NF1 patients (43%).
Chiasmatic OPG. Localization of OPG appears to be an indicator of prognosis (66). Vision is endangered in post-chiasmatic OPG involving the chiasm and hypothalamus. For example, in 62% of patients with post-chiasmatic glioma, reduced vision was noted. On the other hand, only 32% of patients with gliomas confined to the optic nerve and adjacent chiasm suffered from reduced vision (63).
Other authors deny the impact of localization on the predictability of visual functions. They refer to the well-known finding that chiasmatic-post-chiasmatic localized glioma can have an inapparent course (91). Segal et al. (103) support this conclusion. These authors could not prove significant difference between symptomatic and asymptomatic OPG patients with respect to topography of lesion. Following these authors, topography of the space-occupying lesion does not define tumor biology and growth, respectively.
In the present study, the topographical categories ‘chiasmatic’, ‘pre-chiasmatic/chiasmatic’ and ‘chiasmatic/post-chiasmatic’ revealed no significant differences with respect to visual acuity (Table VII).
Nevertheless, patients with symptomatic course were much more frequently affected by PA including the complete optic pathway (10.9% symptomatic OPG vs. 1.6% non-symptomatic OPG with reference to total group; 26.9% symptomatic OPG vs. 2.6% asymptomatic OPGs within the symptomatic and asymptomatic group). Extensive OPG (Dodge III) can indicate an unfavorable course of the disease. However, chiasmatic OPG had no predictive value for the clinical course in this study. Therefore, only trends can be derived from classified localization and prediction of the visual acuity. Dodge grade I have a tendency to show a better course than grade III glioma. However, the factor ‘localization’ is no sufficient parameter to predict prognosis of vision in NF1 patients with OPG.
The course of OPG in patients without therapy. The ophthalmological findings in patients without therapy aged 2 to 18 years mirrors the complete spectrum of the biological tumor behavior of PA. Unilateral OPG did not affect the contralateral eye irrespective of age. On the one hand, unilateral-intraorbitally confined OPG can lead to complete loss of vision of the affected eye. On the other hand, chiasmatic and OPG with post-chiasmatic extension can show a mild course and minimally reduced vision.
Figure 8 shows an increase of vision during the observation period in this group. However, age and compliance of patients have to be considered for interpretation. According to Haase (86) and with reference to Table III, the visual acuity increases rapidly in children and reaches its maximum at the age of 14 to 15 years. With reference to visual acuity, it becomes apparent that there is a substantial difference between ophthalmologically healthy children and those affected with symptomatic OPG in NF1 in all age groups. The median value of visual acuity for the affected (worse) seeing eye of NF1 patients with untreated OPG was 0.4 at the beginning and end of the evaluation period. This is a relevant impairment of vision of the affected eye and a challenging finding for any therapeutic approach. Mean values of visual acuity of the unaffected or only mildly affected eye was around 1.0 at the end of the evaluation period.
The heterogeneous courses of OPG in non-treated patients do not allow prognostic factors such as age of affected individual or localization of the lesion to be derived. Whereas NF1 patients with completely affected optic pathway show a more unfavorable course of disease, the severity of visual impairment dependent on localization is unpredictable in individual cases.
Opocher et al. (124) intended to derive definite prognostic factors for OPG in childhood. They studied 23 publications on this subject, also including PA not associated with NF1. They concluded that age <1 year of life at the time of OPG diagnosis is prognostically relevant and that further prognostic factors such as NF1, tumor site, and others should be discriminated in future studies.
Spontaneous regression of OPG in NF1. Incidence of spontaneous regression of OPG in NF1 is unknown. With reference to published data, the incidence appears to be very low. One study addresses this finding in detail. These authors present their own findings and a review of the literature (120). They diagnosed 3 NF1 patients (2 girls, 1 boy) who had received no therapy and experienced tumor regression, as revealed on serial MRI. Interestingly, during this follow-up, the visual acuity of these patients increased correspondingly. Furthermore, the follow-up of 16 patients with moderate tumor regression was analyzed. Most cases showed a stable vision associated with tumor reduction (n=9), but both visual improvement (n=6) and reduction (n=1) were recorded (55, 56, 121, 122). Volume reduction of OPG is noted independently of NF1 status (57). However, some authors argue that NF1 is an important prognostic factor in spontaneous regression of OPG (120). Our results show that spontaneous regression of OPG is rare, but can be associated with almost complete recovery of vision in the individual case. The short-term cytostatic application is unlikely to have had an impact on the tumor volume in our only case with this course, as depicted on MRI. Occasionally, regressions of OPG were noted following biopsies or partial resection of the tumor in NF1 affected patients (57, 123). Basically, the variable biologic tumor behavior is the limiting factor to determine prognosis in OPG.
The course of OPG in patients with therapy
Chemotherapy. Parameters to indicate chemotherapy in OPG can be: extension of tumor, progressive exophthalmos, reduced vision, optic nerve atrophy, hydrocephalus, endocrine disorders or diencephalic syndrome. Numerous studies on the treatment of OPG have focused on the radiological alterations following chemotherapy, but to a lesser extent on visual acuity (125, 126). These studies often lack a clear distinction between syndromic and sporadic OPG, include a low number of patients for analysis, different evaluation intervals or undescribed primary therapies (68). According to a recent multicenter study (68), the success of chemotherapy in OPG can be summarized like this: vision increases in about every third patient and 22% of all eyes, vision maintains stable in 40% of investigated individuals and 57% of eyes, resp., but gets worse in 28% of patients and 21% of investigated eyes. A relevant conclusion of this analysis of NF1 patients is the stabilization or even improvement of tumor-associated visual deficiency in about one third of patients. A drawback of this study is the lack of a control group consisting of non-treated NF1-OPG patients.
Moreno et al. (127) evaluated 8 publications on this item in their meta-analysis. They considered both sporadic and NF1-associated OPG studies. About 15% of all patients revealed visual improvement and the ophthalmological findings stabilized in 47%. Moreno et al. (127) resumed the majority of patients who were chemotherapeutically treated for glioma will not experience visual improvement. Shofty et al. (128) communicated a similar summary after analysis of sporadic and syndrome associated OPG subjected to chemotherapy with vincristine and carboplatin. During the first chemotherapy phase, allergic reactions were noted after carboplatin and vincristine medication in 7 cases. In 8 cases, no tumor response to the drugs was evident. In total, about 74% of patients experienced an impairment of their vision during the study period.
In the present retrospective study, six out of ten chemotherapeutically treated patients developed severe visual deficiencies (≤0.2) on the affected eye(s) after completion of therapy. It is, however, to be assumed that the marked pre-therapeutic loss of visual acuity in these patients (identified as patients No. C1, C5, C7, C8, C9) has to be considered an irreversible process that could not be halted or reversed with the current chemotherapeutic strategy. Following a visual acuity reduction to 20% or less, the complete restoration of vision is unlikely. Nevertheless, in two (C5, C7) of five patients with severe impaired vision, bilateral residual vision was maintained over the evaluation period (36 and 85 months, resp.). This subgroup consisted of two further cases with a special course of disease. One patient had developed a chiasmatic glioma in early childhood but also developed multiple astrocytomas at other sites. On MRI, these tumors responded to induction therapy. A second patient experienced spontaneous remission after a few applications of chemotherapeutics. In two patients (C2, C4) visual acuity was stabilized at an acceptable level (0.4-0.8).
Radiotherapy. Radiotherapy was in the focus of OPG therapy for a long time. However, these studies lacked the differentiation of sporadic and syndrome associated lesions (129, 130). Ten year progression-free survival of irradiated OPG patients was calculated from 66% to 90% (77, 129). Radiotherapy as the first-choice treatment modality is considered ambiguous in NF1-children with symptomatic OPG. It is important to remember that irradiation exposure of the affected individual can cause malignant transformation of PA and also second primaries (80). Furthermore, in particular children with NF1 aged less than six years are at risk to develop endocrinological damages consequential to irradiation exposure, e.g. growth disturbance or neurological disorders. In cases of no response to chemotherapy for OPG, radiotherapy can be used as a second-line therapeutic option (128). Restraints to applying radiotherapy in this patient group as first-line treatment obviously take note of current publications such as the treatment recommendations according to the brain tumor study (HIT-LGG 1996) published by Gnekow et al. (78). Starting from a total number of 108 NF1 patients, 55 were treated chemotherapeutically and 10 were subjected to radiotherapy (78).
In this study, patients with a history of radiotherapy had a more severe course of disease. In two patients treated after their 18th year of life, unilateral and bilateral complete vision loss was noted. Three further patients could only perceive hand motions at the end of the observation period. On the other hand, one further patient with PA grade I-II showed good vision after radiotherapy on both eyes (1.0). These case reports cannot be taken as being representative for the whole group, but illustrate impressively the variable tumor biology and response to therapy. We could not clarify the possible impact of orbital PNF on vision in 2 patients with respect to the quality of medical files (patient No. R1, R4).
Surgery. Value of surgical biopsy in NF1 associated OPG is controversial. The majority of gliomas are histologically PA WHO grade I. However, astrocytomas of the optic pathway were diagnosed that fulfilled grade III or IV criteria (51). These variations of histological grading are of special interest due to the more aggressive tumor biology in advanced grade PA and the ambiguous effectivity of standard chemotherapy vincristine/carboplatin in these cases. Surgical biopsy is not generally recommended for typical OPG in children but may have some utility in this neoplasm with unusual location or presentation (66).
A further factor to be considered prior to the application of diagnostic surgical intervention is the fact that histological verification and staging of PA cannot be presently used as a basis to assess tumor progression. In this situation the informational value of pathological diagnosis is poor. In the presented study, only four patients were biopsied prior to irradiation. The results were PA grade 1 in three cases and grade 1-2 in one case as expected.
Surgical procedures are taken into consideration to debulk extensive tumor masses and to protect adjacent structures. Furthermore, individual decisions can lead to extensive orbital tumor excisions and even enucleations in cases with progressive exophthalmos and loss of vision (131). Localization and extension are limiting factors for OPG surgery. Complete resection for cerebral LGG is achieved in about 30-95%. Cerebellar LGG are resected in 80-95% (78). Apart from these regions, the percentage of complete resection for LGG is significantly lower in other regions (chiasma opticum, hypothalamus) (78). For example, there is a substantial risk for vascular infarcts in the operation area following subtotal resection for LGG in the chiasm region (132).
Exophthalmos was noted in 6 of 10 surgically treated patients. The finding can be assessed as indicator of tumor progression. In five patients, ipsilateral vision was lost completely after surgery. Nevertheless, progressive exophthalmos is a serious finding in NF1 associated OPG that can require radical surgery. One patient died 27 months after partial tumor resection for hypothalamic extension of OPG due to third ventricle compression. One further patient experienced basal ganglia infarction postoperatively. These results illustrate the value of surgery in advanced stage OPG, the necessity to before and weigh disadvantages of surgery in this identity-forming body region, the inevitable risk for loss of residual vision associated with surgery, and the risk of damaging relevant structures adjacent to the visual pathway. FASI and its importance for NF1 affected individuals. FASI are diagnosed on MRI in 43-93% of NF1-affected children (95, 134). On the other hand, studies on healthy individuals aged 1 to 18 years revealed a prevalence of substantia alba hamartomas in 0.5 to 2.2% (135, 136).
Gill et al. (134) investigated a group of 103 NF1 patients for cerebral anomalies on MRI. They diagnosed signal intense areas in T2-weighted sequences in 66%. Curless et al. (137) revealed FASI using MRI of 284 NF1 affected children (FASI: 57%). A study group of similar size was evaluated by Balestri et al. (94), who reported FASI in 62%. In our study, the differences of FASI frequencies in symptomatic and asymptomatic OPG is low (~6.5%). Patients with symptomatic OPG showed FASI in 49%. It is assumed that there is no correlation between OPG and T2w-hyperintensities on MRI (138). This study provides evidence for the statement that FASI are not predictors of OPG onset in symptomatic or asymptomatic NF1 patients: the distribution of FASI is almost identical in both groups (p=0.942). Our findings are in line with the results of these reports. Ostendorf et al. (97) described the anecdotal finding of T2w hyperintensities that can show radiological alterations during the follow-up, e.g. cystic compartments. They derive from this observation the warning of a present neoplasm that could be mistaken as FASI, in particular in cases with associated neurological findings. In this study, no such relationship was found. The number and size of signal intense areas differed depending on age. It is assumed that the increase of FASI reaches its maximum at age 10 to 12 years and then both parameters decline. Furthermore, FASI were not recorded in children younger than 2 years of age (139, 140). The mean age of patients in this study with FASI was 6.9 and 8.5 years. Both histograms (Figures 15 and 16) confirm the number of FASI reduce with age.
Clinical significance of FASI remains ambiguous. Some authors believe that learning disabilities and minor intelligence quotient values correlate with FASI but other deny this interpretation (110, 141). Some authors recommend the finding of FASI to establish a diagnosis of NF1, in particular in children (142).
Conclusion
Symptomatic OPG in NF1 show an unpredictable course with a wingspan of findings starting on the one side with spontaneous regression and recovery of vision and ending on the other with a fatal course. No clearly predictive factors for OPG could be proven despite a wide range of affected individuals, a sufficiently large number of patients and a thorough analysis of the data. Current discussion on OPG calls into question whether OPG are true neoplasms (32). On the other hand several authors clearly identify OPG as neoplastic entities (131). Slow growth and lack of functional impact of a morphologically defined entity are not accepted parameters to negate a neoplasm. This analysis supports the acceptance of OPG in NF1 as a neoplasm. However, joint interdisciplinary efforts are required to provide more basic science data and clinical experiences in the diagnosis and treatment of OPG in NF1. Furthermore, the numerous ophthalmological findings in NF1 combined with the poor understanding of the pathogenesis of amblyopia should alert clinicians involved in NF1 treatment to refocus on objective methods to study visual acuity in children and to consider additional factors that can have an influence on the vision of these patients (143-153). Avery et al. (70) alert the clinicians caring for children with OPGs to distinguish a decline in visual acuity due to refractive errors (that indicate the need for glasses) from that of OPG-related visual acuity loss. In the context of NF1-affected children, it should be added for consideration that attention deficit disorders may play an important role in determining visual acuity in small children. Attention deficit disorders account up to 50% of children with NF1 (154). In these children, the impact of medication on the visual acuity testing has to be considered, which also varies considerably with the time of the day (VFM, pers. communication).
Recently, the impact of gender on NF phenotype was reiterated (155). We cannot confirm the impact of gender on OPG with respect to our clinical data. Recently, a study group revealed a genotype/phenotype correlation in NF1 patients with OPG (156). However, a consecutive study could not support these results (157).
Reconstructive surgery of the orbital region should consider the visual acuity of NF1 patients with respect to the numerous local neurological and ophthalmological dysfunctions affecting the eye and orbital region. Many of these dysfunctions are inherent to the disease and should be considered a corrective factor of treatment goals in facial reconstructive surgery of the orbital region in NF1 patients. OPG can develop far beyond childhood (158), as already noticed by Lisch (159).
Future work on the diagnosis and treatment of OPG in NF1-affected individuals would benefit from the objectification and wide application of age-adapted visual acuity determination, a uniform definition of inclusion criteria for (drug) therapy, and the publication of long-term follow-up data concerning ophthalmological and neurological findings with respect to therapy (or no therapy). This long-term care of NF1 patients can be provided only in specialized centers.
Acknowledgements
The clinical and radiographic data were kindly provided by Prof. V.-F. Mautner, Neurofibromatosis Outpatient Center, Department of Neurology, UKE. The Authors are grateful for his support during the study and his critical comments on the manuscript. The authors are grateful for the help of Dr. Feucht, formerly senior registrar at the Department of Ophthalmology, UKE, in interpreting the ophthalmological findings of numerous medical sources. Furthermore, Dr. C. Fünsterer, formerly head of the Magnetic Resonance Imaging Center, Hamburg-Othmarschen, supplied the authors with a large number of expert MRI findings.
Footnotes
The results of this study were presented, in part, in oral form on the occasion of 16th International Symposium on Tumor Markers/International Workshop, October, 19th - 21st, 2014, Hamburg, Germany, and 65th Annual Meeting of Maxillofacial Surgery Working Group (DGZMK), May, 14th - 15th, 2015, Bad Homburg v. d. H., Germany.
- Received April 5, 2016.
- Revision received June 9, 2016.
- Accepted June 24, 2016.
- Copyright© 2016 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
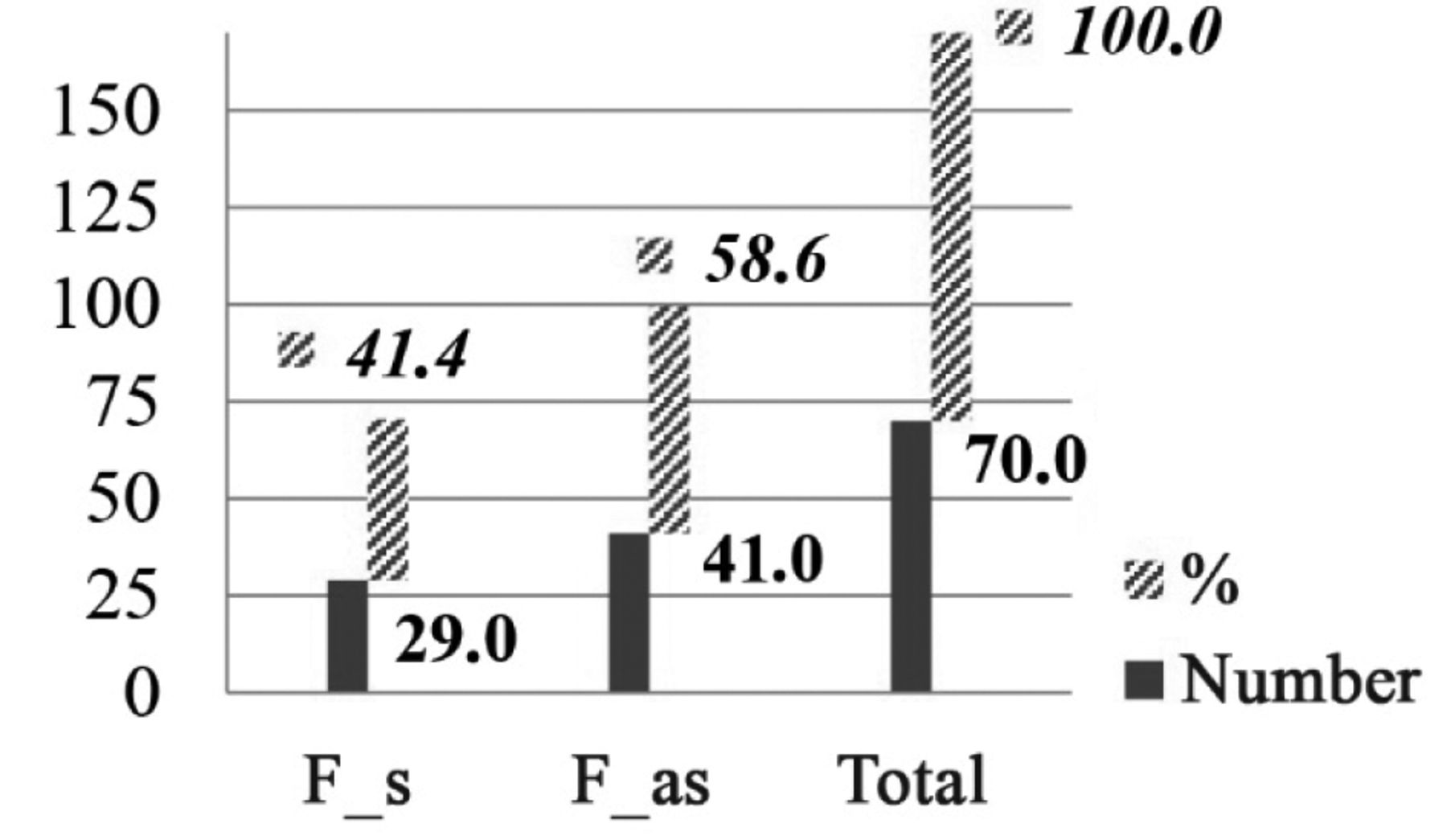{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
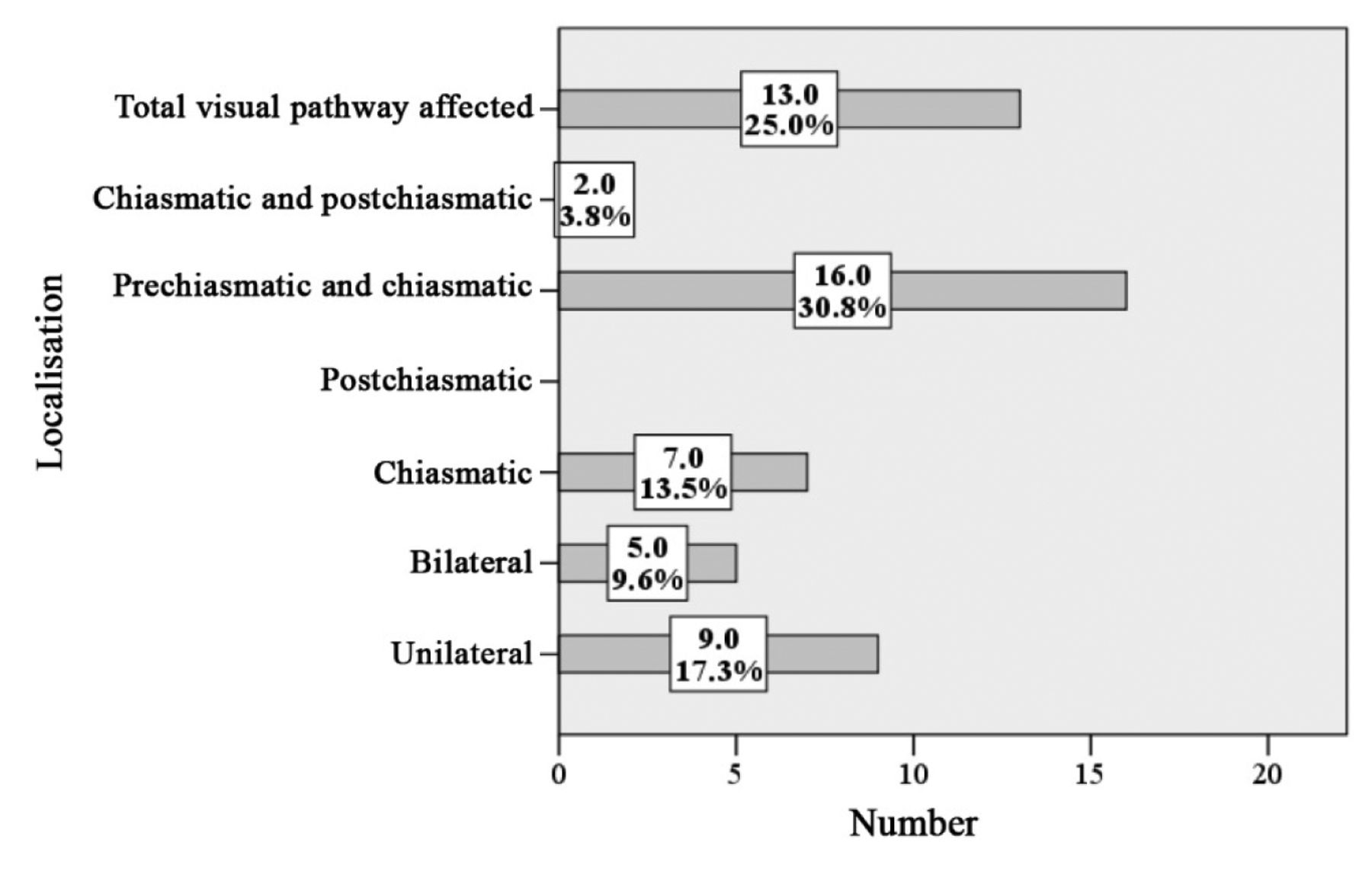{kind=link}
{kind=link}
{kind=link}
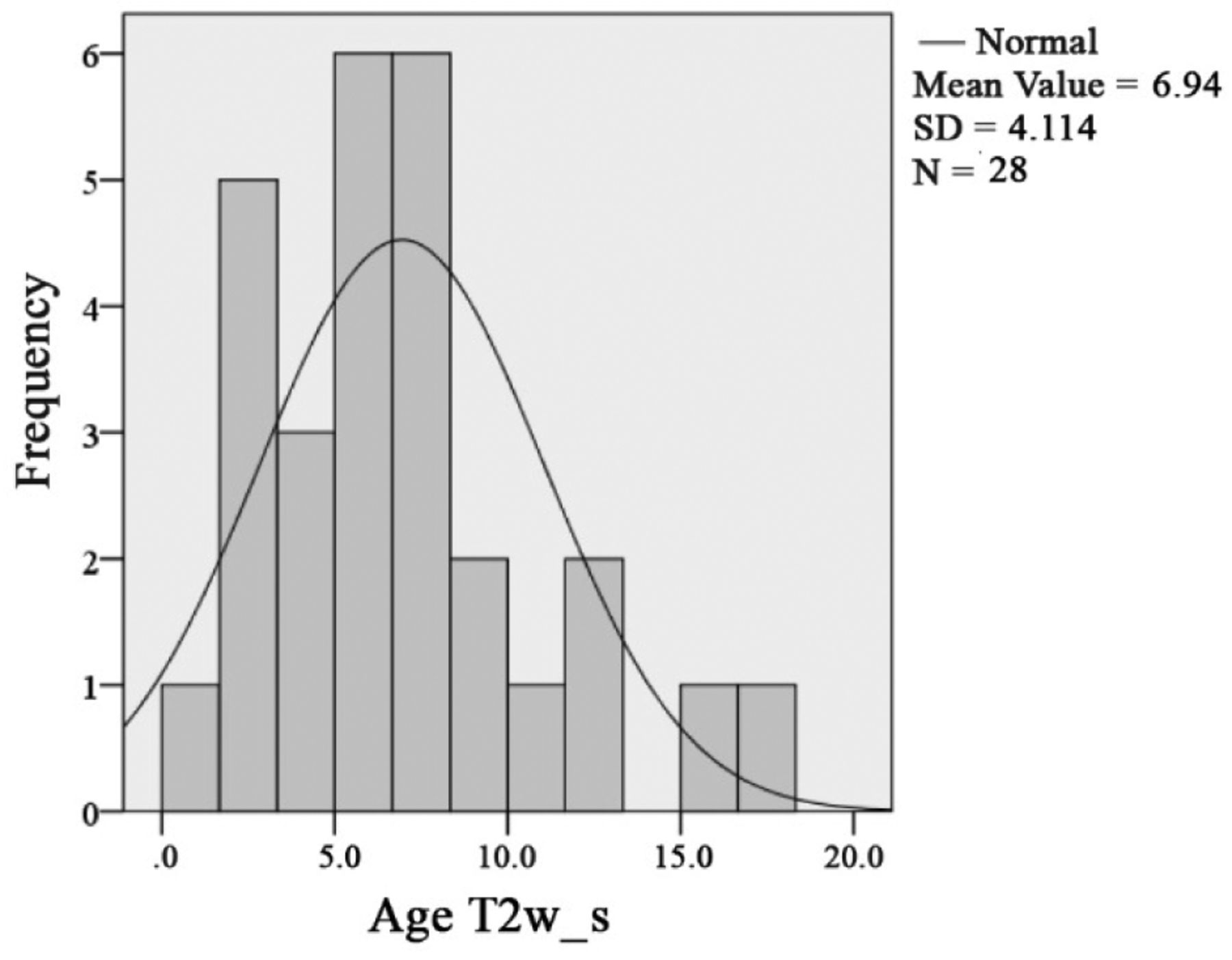{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Peripheral Nerve Sheath Tumors in Patients With Neurofibromatosis Type 1: Morphological and Immunohistochemical Study
- Manifestations and Treatment of Adult-onset Symptomatic Optic Pathway Glioma in Neurofibromatosis Type 1
- Demographical Profile and Spectrum of Multiple Malignancies in Children and Adults with Neurocutaneous Disorders