


Abstract
Background/Aim: Nuclear factor kappa B (NF-κB) inactivation and apoptosis activation have been shown to enhance the anticancer effect of cisplatin in oral squamous cell carcinoma (OSCC). Amentoflavone may suppress NF-κB activity and trigger apoptosis in different types of cancer. The aim of this study was to investigate the anticancer effect and mechanism of amentoflavone in combination with cisplatin in OSCC. Materials and Methods: We investigated the combination effect and mechanism of amentoflavone and cisplatin via cell viability analysis, flow cytometry-based apoptosis analyses, transwell migration/invasion assay, immunofluorescence staining and western blotting assay. Results: Both amentoflavone and QNZ (NF-κB inhibitor) significantly increased cisplatin-induced cytotoxicity. Amentoflavone reduced cisplatin-triggered NF-κB activity and enhanced cisplatin-induced intrinsic caspase-dependent and independent apoptotic pathways. Moreover, amentoflavone augments cisplatin-suppressed invasion and migration ability of OSCC cells. Conclusion: Inactivation of NF-κB and induction of apoptosis through intrinsic caspase-dependent and independent apoptotic pathways are associated with amentoflavone enhanced anti-OSCC efficacy of cisplatin.
Oral squamous cell carcinoma (OSCC) is the most common form of head and neck cancers, which occur in the oral cavity and oropharynx (1). Long-term exposure to carcinogens such as tobaccos, betel nuts and alcohol, is conducive to OSCC formation (2). Cisplatin, which inhibits tumor cell growth by inducing DNA damage, is used to treat patients with OSCC (3, 4). Over 30% of OSCC patients are insensitive to cisplatin treatment. Therefore, development of cisplatin sensitizers may offer therapeutic benefits for OSCC patients treated with cisplatin (5, 6).
Clinical and preclinical studies revealed that combination of cisplatin with other drugs or treatment modalities resulted in better therapeutic outcomes than cisplatin alone or a single modality treatment in OSCC. The combination of postoperative radiotherapy and cisplatin was shown to significantly improve recurrence-free and overall survival compared to radiotherapy alone in OSCC patients (7). For instance, vitamin D and suberoylanilide hydroxamic acid (SAHA, a histone deacetylase inhibitor), have been found to sensitize OSCC to cisplatin through suppression of nuclear factor kappa B (NF-κB) activation (5), as well as to enhance cisplatin-inhibited tumor growth of OSCC by induction of apoptosis (8).
It is anticipated that the combination of NF-κB inactivation and the induction of apoptosis enhance the anti-OSCC efficacy of cisplatin. Amentoflavone (AF), a multifunctional compound isolated from herbal plants, has been presented to inhibit tumor cell growth and invasion through reducing NF-κB signaling and inducing apoptosis in breast cancer, hepatocellular carcinoma, and glioblastoma (9-12). In addition, amentoflavone can enhance sorafenib-induced cytotoxicity through triggering the extrinsic and intrinsic apoptotic pathways of HCC (13). Nevertheless, the ability of amentoflavone to sensitize OSCC to cisplatin is ambiguous. The major purpose of the present study was to investigate the efficacy and mechanism of action of amentoflavone to sensitize OSCC to cisplatin in vitro.
Materials and Methods
Reagents and antibodies. Amentoflavone (AF), cisplatin (CIS), dimethyl sulfoxide (DMSO), Triton X and MTT (3-(4,5-Dimethylthiazol-2-yl-2,5-Diphenyltetrazolium Bromide)) were all purchased from Sigma Aldrich Corp. (St. Louis, MO, USA). Matrigel were purchased from Corning (Tewksbury, MA, USA). BAX, BAK, NF-κB p65 (ser536), NF-κB p65, β-actin, EndoG, AIF were all purchased from (Elabscience, Houston, TX, USA). NF-κB inhibitor (QNZ) 4-N-[2-(4-phenoxyphenyl)ethyl]quinazoline-4,6-diamine (QNZ) was obtained from Selleckchem (Houston, TX, USA).
Cell culture. SAS cells were acquired from Dr. Kai-Wen Hsu’s Lab (China Medical University) and seeded in Dulbecco’s Modified Eagle’s (DMEM) medium containing 10% FBS and 1% Penicillin-Streptomycin (Penicillin G Sodium Salt: 10,000 units/ml; Streptomycin Sulfate: 10 mg/ml). SAS cells were incubated in a 37°C incubator under a humidified 5% CO2 atmosphere. Cell culture related products were all obtained from Thermo Fisher Scientific Inc., MA, USA.
Cell morphology, cell viability and combination index analyses. SAS cells were seeded as 3×104 cells/well in 96-well plate and incubated overnight. Cells were treated with amentoflavone (0, 100, 150, 200, 300 μM), QNZ (0.5 μM) and cisplatin (0-5 μg/ml) for 24 h. Cell morphology was analyzed with a Nikon ECLIPSE Ti-U microscope (Minato City, Tokyo, Japan). MTT assay was used for cell viability analysis. After treatment of cells with the MTT reagent (stock 5 mg/ml, diluted ten times with medium), cells were incubated in a 37°C incubator for 4 h. Living cells were detected by a SpectraMax iD3 microplate reader (Molecular Devices, San Jose, CA, USA), as previously described (14). The combination index (CI) analysis was used to identify suitable combination dosage. The CI value calculation was performed by the CompuSyn software (ComboSyn, Paramus, NJ, USA), which was developed by Chou and Talalay (15).
Mitochondria membrane potential (ΔΨm) analysis. SAS cells were seeded as 5×105 cells/well in a 6-well plate for overnight incubation. Cells were treated with 100 μM amentoflavone and 1 μg/ml cisplatin for 48 h. After treatment, cells were harvested and stained with 4 μM 3,3’-dihexyloxacarbocyanine iodide (DiOC6) dye for 30 min in the dark (Thermo Fisher Scientific). After staining, fluorescence intensity from cells were evaluated by NovoCyte flow cytometry. The results of the staining were measured using NovoExpress® software (Agilent Technologies Inc., Santa Clara, CA, USA).
Cell cycle subG1 phase and annexin-V/PI analyses. SAS cells were seeded as 5×105 cells/well in 6-well plate for overnight incubation. Cells were treated with 100 μM amentoflavone and 1 μg/ml cisplatin for 48 h. For cell cycle subG1 analysis, cells were harvested and fixed with 75% ethanol overnight at –20°C. The next day, cells were stained with PI solution (for cell cycle analysis, 40 μg/ml PI, 100 μg/ml RNase and 1% Triton X-100 in PBS) for 30 min at 37°C. For annexin-V/PI analysis, cells were harvested and stained with Annexin-V/FTC apoptosis detection kit (Vazyme Biotech Co. Lt, Nanjing City, PR China) in the dark for 30 min at 37°C. After different staining procedures, apoptosis-related signals in cells were finally evaluated by NovoCyte flow cytometry and quantified by NovoExpress® software (16).
Caspase-3, caspase-8 and caspase-9 analyses. SAS cells were seeded at a density of 5×105 cells/well in 6-well plates for overnight incubation. Cells were treated with 100 μM amentoflavone and 1 μg/ml cisplatin for 48 h. CaspGLOW™ Fluorescein Cleaved Caspase-3, CaspGLOW™ Fluorescein Cleaved Caspase-8, and CaspGLOW™ Fluorescein Cleaved Caspase-9 staining kit were purchased from Biovision (Mountain View, CA, USA). For activation of caspase-3, caspase-8 and caspase-9, the staining procedure was performed according to the manufacturer’s protocol and as previously described (17, 18).
Invasion and migration transwell assay. SAS cells were seeded at a density of 3×106 cells/well in 10 cm dishes for overnight incubation. Invasion and migration analyses were performed by the Corning 8 μm transwell system (with or without matrigel) as previously described (18). Membranes from transwells were stained, photographed using the Nikon ECLIPSE Ti-U microscope and quantified by ImageJ software (version 1.50, National Institutes of Health, Bethesda, MD, USA) (18).
Western blot. SAS cells were seeded at a density of 3×106 cells/well in 10 cm dishes for overnight incubation. Cells were treated with 100 μM amentoflavone and 1 μg/ml cisplatin for 48 h. Total protein was harvested, separated by 8-12% SDS pages, and transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were then incubated with primary antibodies, including BAX, BAK, NF-κB p65 (Ser536), NF-κB p65 and β-actin (Cell Signaling Technology, Danvers, MA, USA). Detailed procedure is described in a previous study (19).
Immunofluorescence (IF) staining assay. SAS cells were seeded at a density of 2.5×104 cells/well in a 4-well chamber slice overnight (Nunc™ Lab-Tek™ II Chamber slide™, Thermo Fisher Scientific, Fremont, CA, USA). Cells were treated with 100 μM amentoflavone and 1 μg/ml cisplatin for 48 h. After treatment, cells were fixed with 4% paraformaldehyde for 15 min, permineralized with 0.1% Trtiton-X-100, washed 3 times with PBST (phosphate-buffered saline, 0.1% Tween-20), and blocked with blocking buffer (1% BSA) for 1 h at 25°C. Cells were then incubated with a primary antibody in blocking buffer (1:300) overnight at 4°C. After 3 washes, cells were stained with Alexa Fluor 488 conjugated secondary antibody (1:400) in blocking buffer for 1h in the dark. Finally, chamber slices were covered by cover slips (24×50 mm) and DAPI mounting medium. IF staining slides were photographed by Zeiss Axio Scope.A1 fluorescence microscope.
Statistical analysis. Significant difference between groups was analyzed by one-way ANOVA using Microsoft excel 2017 version. p-Values less than 0.05 were considered as statistically significant. Data are presented as mean±standard deviation.
Results
The promotion of cisplatin-induced cytotoxicity by amentoflavone was associated with NF-κB inhibition in OSCC cells. As showed in Figure 1A, SAS cells viability was decreased by amentoflavone in a dose-dependent manner. Then, we identified whether the inhibition of NF-κB by QNZ induces the cytotoxicity of cisplatin. As illustrated in Figure 1B, viability was markedly reduced after treatment with QNZ combined with cisplatin as compared to cisplatin treatment alone. Therefore, we aimed to identify whether amentoflavone acts through NF-κB to enhance the toxicity of cisplatin in OSCC. The combination of amentoflavone with cisplatin displayed superior cytotoxicity (Figure 1C). Then, we calculated the suitable combination dosage to obtain a synergistic effect with amentoflavone and cisplatin. As indicated by the CI value (CI=0.82), 100 μM of amentoflavone combined with 1 μg/ml of cisplatin may synergistically decrease cells viability (Figure 1D and Table I). Bright field images of SAS cells obtained with a microscope, displayed a decreased density and cells shrinkage after the combined treatment (Figure 1E). Furthermore, the phosphorylation of NF-κB induced by cisplatin was diminished by amentoflavone (Figure 1F). In summary, we suggested that the cytotoxicity of cisplatin can be enhanced by amentoflavone via NF-κB inactivation.
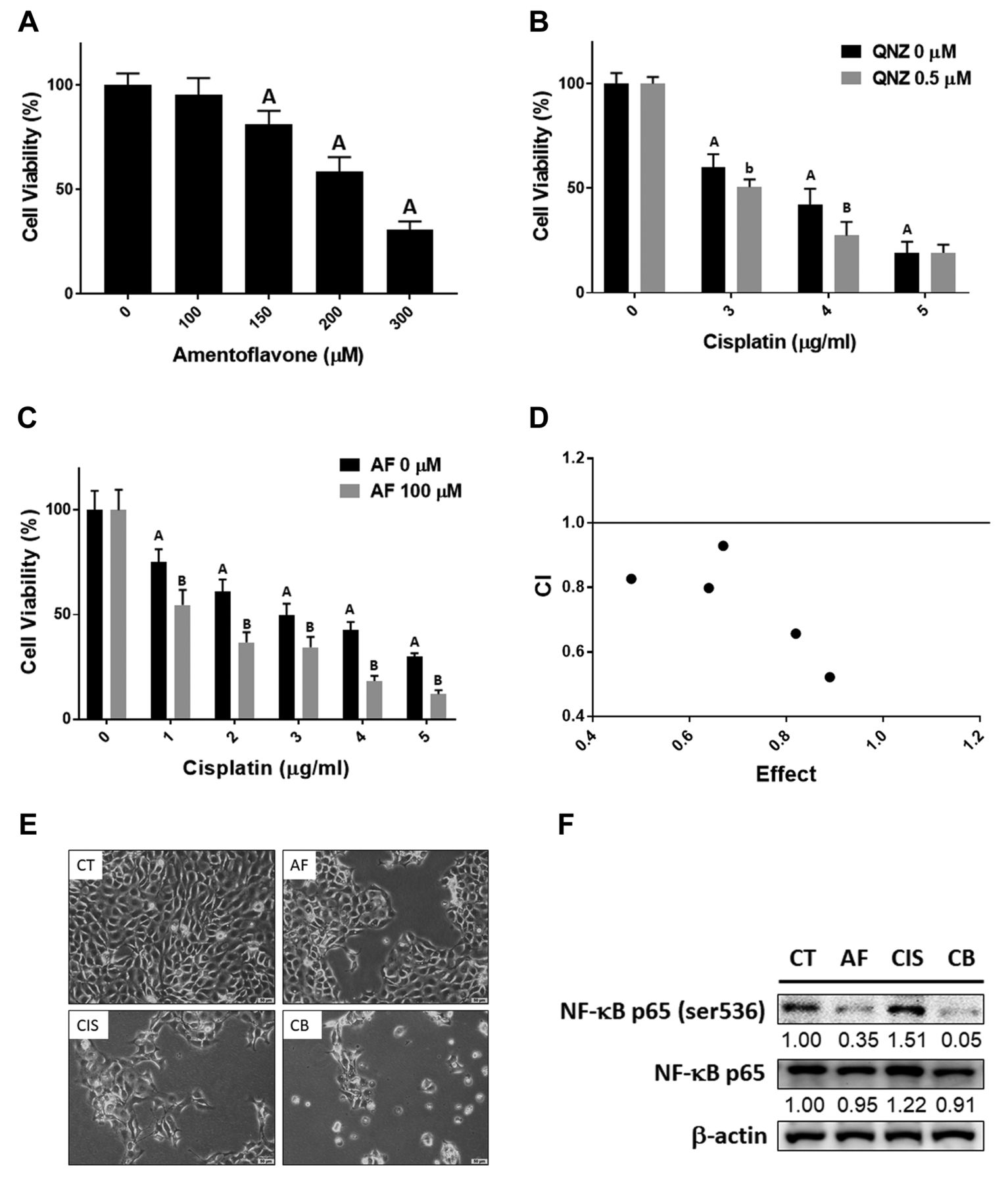
Cytotoxicity effect of amentoflavone and cisplatin in SAS cells. MTT assay results of (A) amentoflavone alone, (B) cisplatin combined w/o 0.5 μM QNZ and (C) cisplatin combined w/o 100 μM amentoflavone treatment for 24 h in SAS cells. (D) The CI value of the combination treatment. (E) The morphology and density change of SAS cells after amentoflavone, cisplatin and combined treatment. (F) The protein expression of NF-κB after treatment as analysed by Western blotting. Scale bar=50 μm. Ap-value<0.01 vs. non-treated group; bp-value <0.05, Bp-value <0.01 vs. single treatment group; AF: amentoflavone; CIS: cisplatin; CB: combination.
Combination index value of amentoflavone combined with cisplatin. The CI value calculation was performed by CompuSyn software (ComboSyn, Paramus, NJ, USA). The combination effect was defined as follow, synergism (CI<1 ), additive Effect (CI=1) and antagonism (CI>1 ).
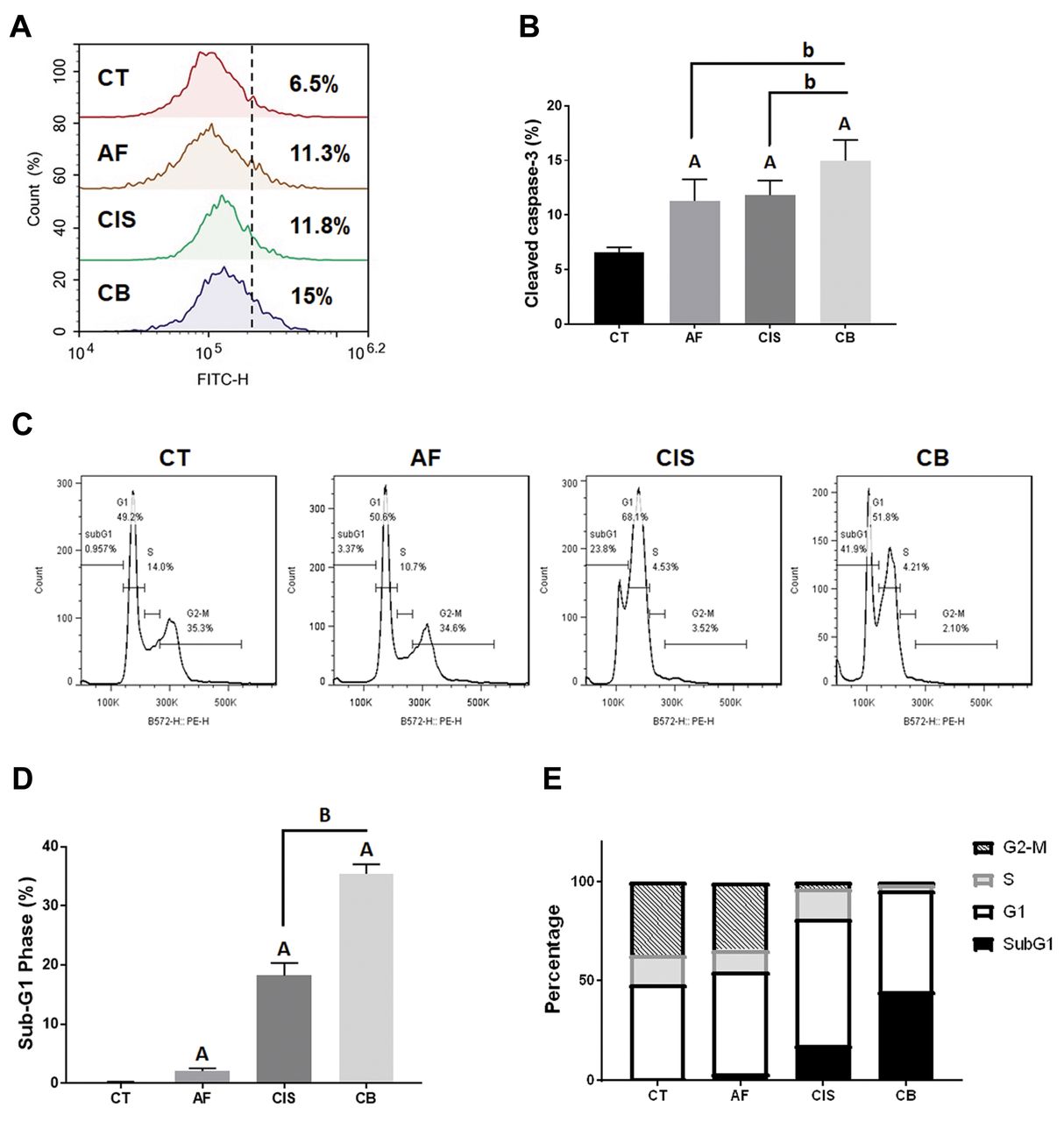
Amentoflavone enhances cisplatin-induced apoptosis in OSCC cells. To identify the apoptosis effect of amentoflavone combined with cisplatin, we investigated the expression levels of several apoptosis-related markers. As shown in Figure 2A and B, cleaved caspase-3 was activated by the combination of amentoflavone with cisplatin. The percentage of cells in the subG1 phase increased after treatment with cisplatin and was further increased after treatment with amentoflavone combined with cisplatin (Figure 2C and D). Furthermore, the percentage of cells in the G1 phase increased after treatment with cisplatin alone or the combination (Figure 2E).

Apoptosis effect of amentoflavone and cisplatin in SAS cells. (A-B) The activation of cleaved-caspase-3 and (C-D) the accumulation of cells in the subG1 phase after treatment are displayed. (E) The quantification of cells in the different phases of the cell cycle after treatment. Ap-value<0.01 vs. non-treated group; bp-value <0.05, Bp-value <0.01 vs. single treatment group.
Amentoflavone enhances cisplatin-induced intrinsic caspase-dependent and independent apoptosis in OSCC cells. After confirming the apoptosis effect of amentoflavone combined with cisplatin, we further aimed to identify which apoptotic pathway was activated by the combination treatment. A loss of the mitochondrial membrane potential (ΔΨm) was induced by the combination treatment (Figure 3A and B). Furthermore, treatment with the combination of amentoflavone and cisplatin resulted in the induction of BAK and BAX (Figure 3C) as well as the activation of cleaved caspase-9 (Figure 3D). Thus, the intrinsic caspase-dependent apoptotic pathway was activated by the combination treatment. However, as shown in the caspase-8 analysis, the combination of amentoflavone with cisplatin may not effectively activate the extrinsic apoptotic signaling (Figure 3E). In addition, the activation of AIF and EndoG was confirmed by IF staining, which indicated also the activation of a caspase-independent apoptosis signaling (20, 21). As illustrated in Figure 3F and G, AIF and EndoG nuclear translocation was increased in the combination group as compared to the single treatment. In summary, amentoflavone may enhance cisplatin induced both intrinsic caspase-dependent and independent apoptosis pathway in OSCC.

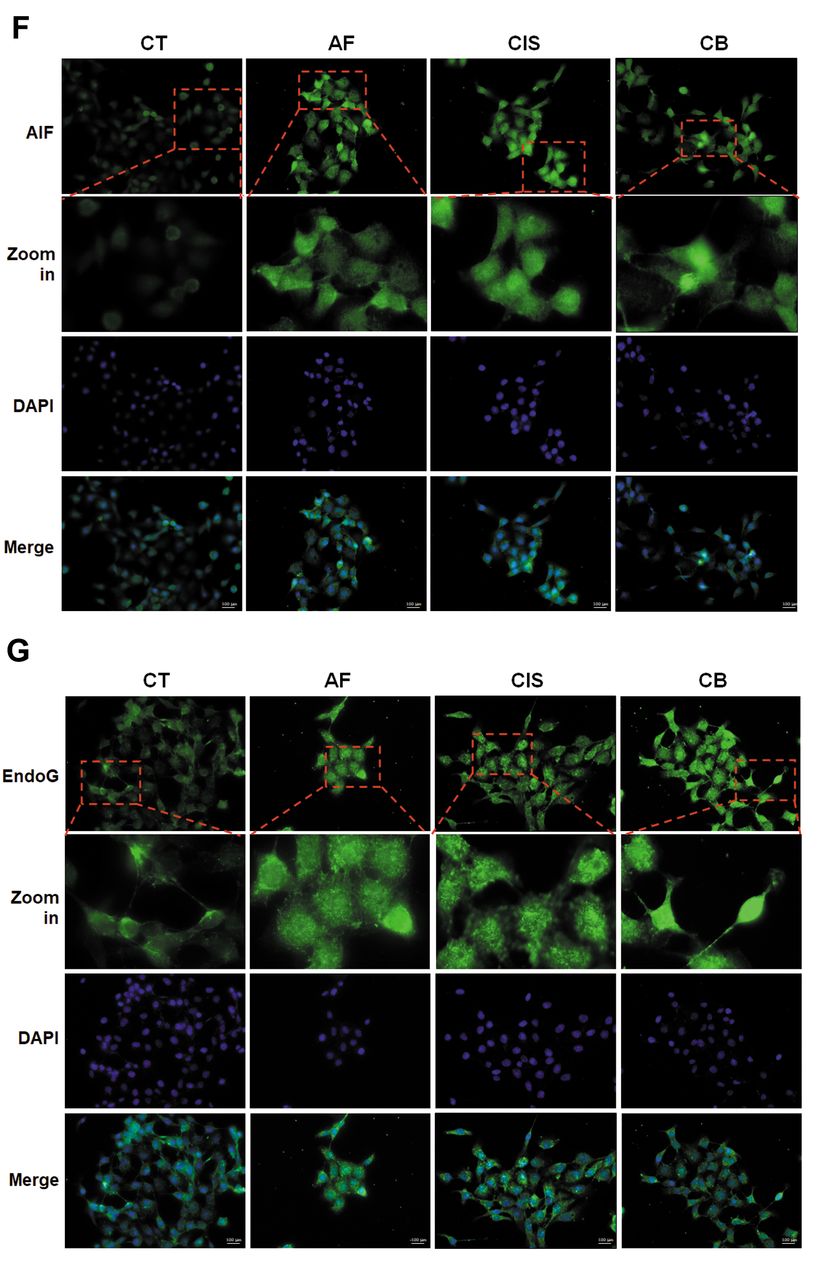
Intrinsic caspase-dependent and caspase-independent apoptosis were induced by amentoflavone combined with cisplatin in SAS cells. (A-B) The loss of ΔΨm after single and combined treatment is displayed. (C) The protein expression of BAX and BAK after single and combined treatment is displayed. The activation of (D) cleaved caspase-9 and (E) cleaved caspase-8 after single and combined treatment are displayed. The IF staining results of (F) AIF and (G) EndoG after single and combined treatment are displayed. ap-value<0.05, Ap-value<0.01 vs. non-treated group; Bp-value<0.01 vs. single treatment group.
Amentoflavone enhances the cisplatin-induced suppression of invasion and migration of OSCC cells. To identify whether invasion and migration may be further reduced by amentoflavone combined with cisplatin, we performed invasion/migration transwell assay. As indicated in Figure 4A and B, the percentage of cells that migrated was decreased in the combination group as compared to the single treatment groups. In addition, the number of invaded cells was also further reduced by the combined treatment as compared to the single treatment groups (Figure 4A and C).

Migration and invasion effect were inhibited by amentoflavone combined with cisplatin in SAS cells. (A) The invasion and migration pattern on the transwell membrane are displayed. (B-C) The invasion and migration percentage of cells in each group are quantified and displayed. Ap-value<0.01 vs. non-treated group; bp-value<0.05, Bp-value<0.01 vs. single treatment group; scale bar = 100 μm.
Discussion
NF-κB, an oncogenic transcription factor, is known to promote cell growth, survival, angiogenesis, epithelial-mesenchymal transition, and invasion through upregulating the expression of NF-κB target genes in cancers (22-25). Constitutive NF-κB activity is associated with chemoresistance and tumor metastasis in OSCC patients (26, 27). Cisplatin triggers NF-κB activity, which in turn results in resistance of OSCC to cisplatin. Therefore, inhibition of NF-κB has been used to sensitize OSCC to cisplatin (15, 28). Our results showed that QNZ (a specific NF-κB inhibitor) and amentoflavone increase cisplatin-induced cytotoxicity and reduce cisplatin-triggered NF-κB activity, respectively (Figure 1B and F). In addition, amentoflavone significantly enhanced cisplatin-mediated inhibition of cell proliferation, migration, and invasion of SAS cells (Figures 1C and 4).
Inhibition of tumor growth can be induced by anticancer agents through caspase-dependent and independent apoptotic pathways (29-31). During the initiation of the extrinsic and intrinsic apoptotic pathways, cleaved-caspase-8 and 9 participate in the regulation of DNA fragmentation via cleavage of caspase-3 (32, 33). Intrinsically, the proapoptotic proteins BAX and BAK, initiate loss of the mitochondrial membrane potential (ΔΨm) and result in the release of cytochrome-c, apoptosis-inducing factor (AIF) and endonuclease G (EndoG) from mitochondria. Both AIF and endoG are caspase-independent death effectors that translocate to the nucleus and induce apoptotic DNA fragmentation (34-36).
In this study, amentoflavone augmented the cisplatin-induced apoptotic signaling. The combination of amentoflavone and cisplatin significantly increased expression of cleaved caspase-3 and induced cell accumulation in the sub-G1 phase, compared to amentoflavone or cisplatin single treatment (Figure 2). Different from the extrinsic and intrinsic apoptotic signaling triggered by the combination of amentoflavone and sorafenib in HCC (13), our results indicate that amentoflavone significantly enhanced cisplatin-induced loss of ΔΨm and caspase-9 cleavage, but not caspase-8 cleavage in SAS cells (Figure 3A, B, D and E). In addition, the combination of amentoflavone and cisplatin further upregulated BAX and BAK expression compared to that induced by amentoflavone or cisplatin alone. The intrinsic apoptotic pathway, which is characterized by increased expression of BAX or BAK, was found to correlate with a favorable prognosis of OSCC patients (37). Notably, amentoflavone also intensified cisplatin-triggered AIF and EndoG nuclear translocation (Figure 3F and G).
In conclusion, the anti-OSCC effects elicited by co-treatment of amentoflavone with cisplatin are at least partly caused by NF-κB inactivation as well as by the induction of apoptosis through intrinsic caspase-dependent and independent pathways. With regard to the antitumor efficacy and sensitization effect of amentoflavone, we suggest that amentoflavone may cause therapeutic benefits to OSCC patients treated with cisplatin.
Acknowledgements
Experiments and data analysis were performed in part through the use of the Medical Research Core Facilities Center, Office of Research & Development at China Medical University, Taichung, Taiwan.
Footnotes
↵*† These Authors contributed equally to this study.
Authors’ Contributions
Data curation, CH Chen, YC Huang, ZL Tan, and FT Hsu; funding acquisition, CH Chen and FT Hsu; writing—original draft, HF Tu, CJ Tsai, YC Chuang, YJ Lee and FT Hsu; writing—review, HF Tu, TC Liu and FT Hsu. All Authors have read and agreed to the published version of the manuscript.
This article is freely accessible online.
Conflicts of Interest
The Authors declare that they have no conflicts of interest in relation to this article.
Funding
This study was supported by China Medical University, Taichung, Taiwan (grant number: CMU109-MF-07), Show Chwan Memorial Hospital, Changhua, Taiwan (Grant number: SRD-109002), National Yang-Ming University Hospital, Yilan, Taiwan (Grant number: RD2019-002), Ministry of Science and Technology, Taipei, Taiwan (Grant number: MOST 108-2314-B-010-014), respectively. This work was also financially supported by the “Drug Development Center, China Medical University” from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan.
- Received October 9, 2020.
- Revision received October 19, 2020.
- Accepted October 20, 2020.
- Copyright © 2020 International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}