


Abstract
Background/Aim: Neurocutaneous disorders, also referred as phacomatoses, are congenital disorders manifesting at different ages with central nervous system and cutaneous abnormalities. Analysis of the demographic and clinical profile of patients with phacomatoses in the context of the incidence and spectrum of malignancy. Materials and Methods: This is a retrospective analysis of 20 years of data in a single-center study in Poland. Results: Phacomatoses were diagnosed in 45.6% (913/2,003) of referred patients, including 61.4% children. The distribution of phacomatoses included: neurofibromatosis type 1 (NF1) in 92.4%, tuberous sclerosis complex (TSC) 3.9%, neurofibromatosis type 2 (NF2) 2.0%, Klippel-Trenaunay syndrome 0.5%, Von Hippel-Lindau syndrome 0.5%, and other sporadic diseases 0.7%. Non-phacomatosis patients were diagnosed mainly for cafe-au-lait-macules (42.8%). The frequency of malignancy was 9.4% (86/913), including 9.1% in patients with NF1; 27.8% in NF2; and 8.3% in TSC. Multiple malignancies were diagnosed in 0.7% and 7% of all phacomatosis and malignancy-diagnosed patients, respectively. Conclusion: The risk of malignancy in patients with phacomatoses was 21.3-fold higher than in the general population. The risk of secondary malignancy was 7%.
Neurocutaneous disorders, also referred to as phacomatoses, are congenital disorders manifesting at different ages with central nervous system and cutaneous abnormalities. These disorders are regarded as rare diseases, which are defined by an incidence below 1:2,000 individuals in the general population. More than 30 entities form this group including the most frequent disorders: neurofibromatosis types 1 (NF1) and 2 (NF2), von Hippel-Lindau syndrome (vHLS), tuberous sclerosis complex (TSC), Klippel-Trenaunay syndrome (KTS) and others (1). Each of these conditions is caused by distinct genetic defects, and the disorders share as pathogenetic mechanism mutations of tumor suppressor genes. These genetic abnormalities attribute high susceptibility to the development of benign and malignant tumors (2).
In Poland, which is a country with 38 million inhabitants, the Phacomatosis Center in Bydgoszcz is the largest, and provides health-care both for children and adults with neurocutaneous disorders from northern, western and eastern part of the country. The objective of this study was the analysis of the demographic and clinical profile of patients with neurocutaneous disorders in the context of the incidence, the type and the spectrum of malignancy.
Patients and Methods
Data collection. The medical records of patients suspected and diagnosed for neurocutaneous disorders who were followed-up in the Phacomatosis Center in the Department of Pediatric Hematology and Oncology in Bydgoszcz from 1999 to 2018 were analyzed. Medical records of patients were retrospectively reviewed for demographic and clinical data including occurrence of malignancies.
Criteria for clinical diagnosis of phacomatoses. Neurofibromatosis type 1 (NF1): It is diagnosed in patients with ≥2 of the 7 following features: six or more cafe au lait macules (CALMs) (>0.5 cm in diameter before puberty and >1.5 cm in diameter after puberty), axillary and/or inguinal freckling, Lisch nodules (iris hamartomas), two or more discrete neurofibromas or one or more plexiform neurofibromas, an optic pathway glioma (OPG), distinctive bony lesions (i.e., orbital dysplasia and long bone dysplasia), and a first-degree relative with NF1 (3, 4). NF1 is an autosomal-dominant disorder, caused by changes in the NF1 gene (17q11.2). NF1 gene encodes neurofibromin 1 protein, expressing a tumor suppressor function (5).
Neurofibromatosis type 2 (NF2). It is a tumor-prone disorder characterized by the development of multiple schwannomas, meningiomas and ependymomas, with frequent hearing loss. NF2 is a microdeletion syndrome involving mutations in the NF2 gene (22q12.2) (6).
Von Hippel-Lindau syndrome (vHLS). Clinical hallmarks of vHLS include the development of retinal and central nervous system (CNS) hemangioblastomas (blood vessel tumors), pheochromocytomas, multiple cysts in the pancreas and kidneys, and an increased risk for malignant transformation of renal cysts into renal cell carcinoma. More than 370 inherited mutations in the VHL gene (3p25.3) have been identified in people with vHLS (6).
Tuberous sclerosis complex (TSC). It is a disorder characterized by hamartomas and malignant tumors in different body organs, mainly in the brain, skin, kidney, liver, lung, and heart. The clinical manifestations of TSC are the result of a mutation in one of two tumor suppressor genes, TSC1 (9q34) and TSC2 (16p13.3) (7).
Klippel-Trenaunay syndrome (KTS). It is marked by a port-wine stain (a red or purplish birthmark caused by swelling of small blood vessels near the surface of the skin), abnormal overgrowth of bone and soft tissue, and varicose veins or other abnormal veins and lymph vessels. It usually affects the legs, but it can also affect the arms or trunk. KTS can be caused by mutations in the PIK3CA gene (3q26.32) (8).
Statistical analysis. Data were expressed as the means and standard deviations or the medians and ranges, depending on the parametric or nonparametric distribution of the variable. Baseline parameters were compared between both groups using Mann–Whitney tests for quantitative variables, and Chi-square or Fisher's exact tests for frequencies. Odds ratio (OR) and 95% confidence intervals (95%CI) were calculated for the differences in frequencies between the groups. Cumulative incidence of age-dependent diagnosis was calculated by Kaplan–Meier method and compared with log-rank test. Data analysis was carried out using SPSS 25 (IBM SPSS Statistics, Version 25.0). Statistical significance was regarded as p<0.05.
Results
Demographics. Total number of 2003 patients suspected for neurocutaneous disorders were referred to the Phacomatosis Center between 1999 to 2018, including 1030 females (51.4%) and 973 males (48.6%), with a median age of 9.8 years (range=23 days-79 years). Overall, 913 (45.6%) patients (males 415, i.e. 45.5%, and females 498, i.e. 54.5%) at a median age of 14 years, met the diagnostic criteria of phacomatoses, including 561 (61.4%) children and 352 (38.6%) adults. A total of 1,090 (54.4%) were not diagnosed for phacomatoses and remained the as observational group.
Clinical profile. Patients with a NF1 diagnosis (843/2003, i.e. 42.1%) were the predominant group in the cohort. Seventy (3.5%) patients were diagnosed for other, rare neurocutaneous disorders: TSC, NF2, KTS, VHLS and other sporadic diseases (hypomelanosis Ito, Rendu-Osler-Weber syndrome, Peutz-Jaghers syndrome, Noonan syndrome, Leopard syndrome and Christ-Siemens-Touraine syndrome) (Figure 1).
Patients not diagnosed for phacomatoses included mainly those who were suspected for NF1 due to CALM (42.8%). Other causes of patient's referral included: skin macules (3.7%), hemangiomas (3%), vitiligo spots (1.4%), hemihypertrophy (0.3%), relatives of patients with phacomatosis (0.7%), and other rare causes (2.5%).
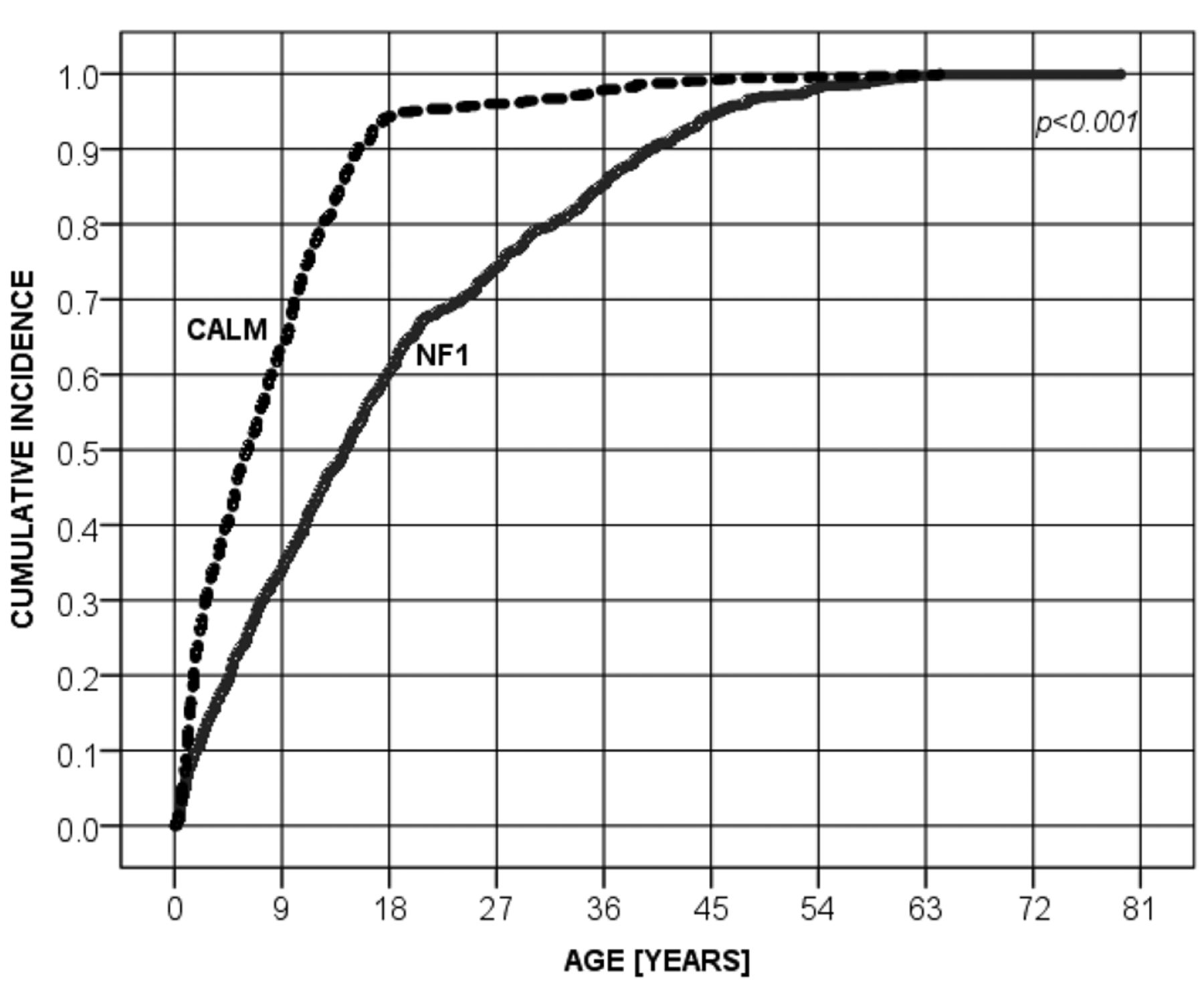
The median age of patients with CALM was significantly lower than that of patients diagnosed with NF1 (6.1 vs. 14.2 years) (p<0.0001) (Figure 2). Still, 329/843 (39.5%) patients with NF1 were diagnosed in adulthood.
NF1 patients were diagnosed to have plexiform neurofibromas in 16% (135/843) and optic pathway gliomas (OPG) in 10.2% (86/843) cases. Plexiform neurofibromas were observed in 60 (44.4%) males and 75 (55.6%) females, at the median age of 18.7 years. OPG were found in 52 (64.7%) males and 34 (39.5%) females, at the median age of 6.6 years.
Malignancies. The frequency of a malignancy occurring in patients diagnosed with neurocutaneous disorders was 9.4% (86/913). With respect to primary diagnosis, the frequency of malignancy was 9.1% (76/843) in patients with NF1; 27.8% (5/18) in NF2 (p=0.007; OR=3.8, 95%CI=1.3-11); 8.3% (3/36) in TSC (ns); 20% (1/5) in vHLS and 20% (1/5) in KTS (ns). In this group, 6/86 (7%) patients were diagnosed with more than one cancer including two patients with three malignancies (Table I). The risk of multiple malignancies among all patients with neurocutaneous disorders was 0.7% (6/913). Overall, the total number of malignant tumors observed among all patients was 94. The most common types of malignancies were brain tumors (46/94; 53.5%, including 30%, 14/46 with OPG), malignant peripheral nerve sheath tumor (MPNST),progressive plexiform neurofibromas (17/94; 18.1%), breast cancers (7/94; 7.4%), sarcomas (7/94; 7.4%), adrenal tumors (4/94; 4.3%), thyroid cancers (2/94; 2.1%), melanomas (2/94; 2.1%), renal tumors (2/94; 2.1%), liver tumor (1/94; 1.1%), endometrial cancer (1/94; 1.1%), colorectal carcinoma (1/94; 1.1%), nasopharyngeal carcinoma (1/94; 1.1%), GIST (1/94; 1.1%), and schwannoma malignum (1/94; 1.1%). One patient developed aplastic anemia (1/94; 1.1%).
Comparison with the general population. With the estimated 0.43% (163,281/37.95 million) incidence of malignant diseases in the general Polish population in the year 2015 (9), the risk of a malignancy in our large group of patients with phacomatoses was 21.3-fold (95%CI=16.7-30.3) higher than in the general population.
Discussion
There is an association between neurocutaneous disorders and development of malignancies. In our study, the frequency of malignancy in patients with phacomatoses was 9.4%. Patients with NF1 diagnosis were the predominant group in this population, appearing in 76/86 (88.4%) patients with malignant diseases. As in other tumor-predisposing syndromes, NF1-associated malignancies generally occur at an early age and often have a neural crest and myeloid origin (2, 10).
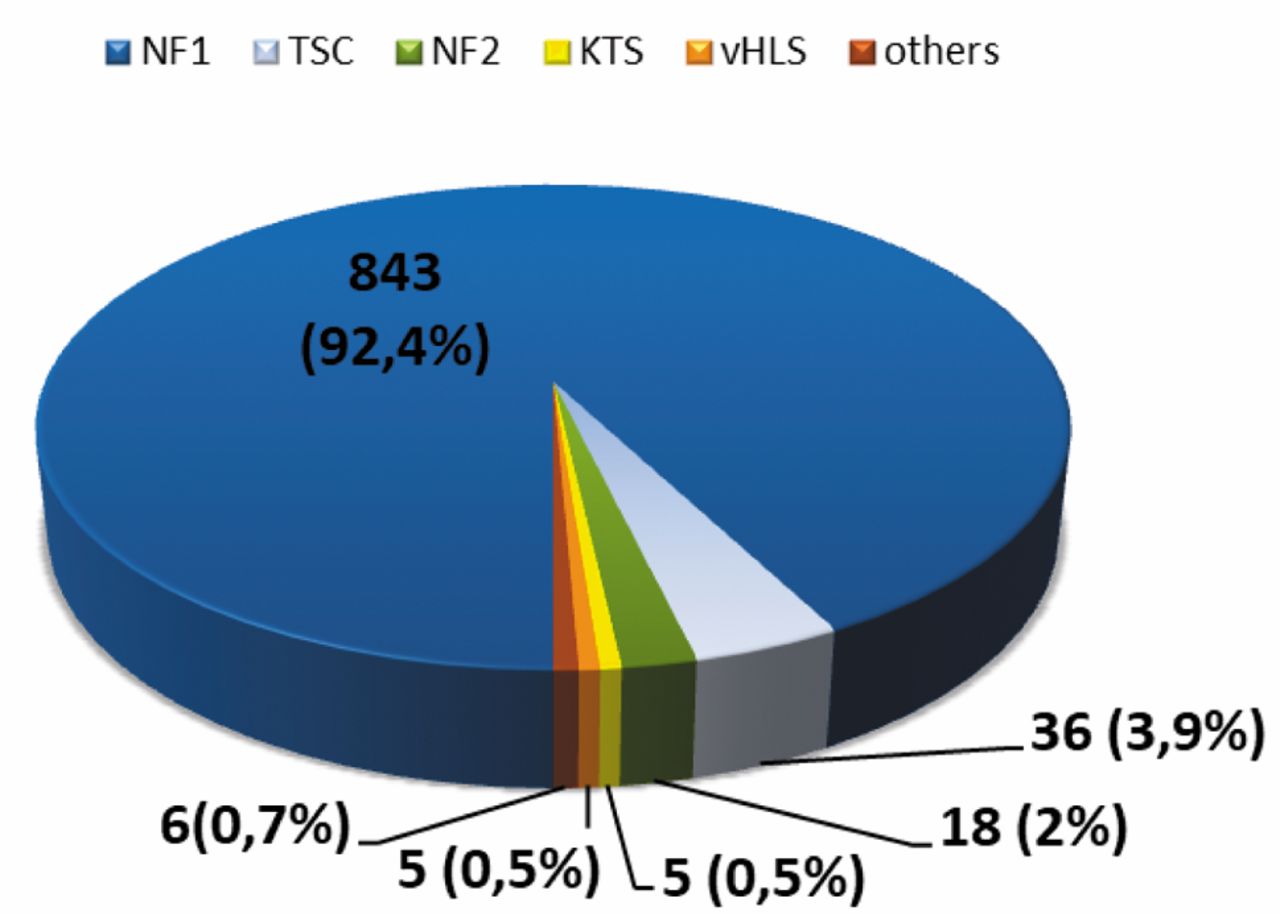
The distribution of patients with neurocutaneous disorders.

Age at diagnosis of NF1 and CALM.
Characteristics of patients with multiple malignancies.
Neurofibromas are the most common tumor in NF1, and plexiform neurofibromas are considered to be pathognomonic lesions of the disease and occur in about 30% of affected individuals (10). Plexiform neurofibromas were observed in 16% (135/843) NF1-patients in our cohort, and in 12.6% (17/135) they gave rise to MPNST, that contributed to 18.1% (17/94) of the total number of malignancies. In other studies, the risk of developing MPNST in NF1-affected individuals has been estimated at 8-13%, compared to 0.001% in the general population (11).
OPG is the second most common tumor in NF1 observed in about 15% of NF1 patients. It can be found at any age, but most frequently is occurring by the sixth year of age (4, 10). In our cohort, about 10.2% (86/843) of individuals had OPGs. The median age of patients with OPG was 6.6 years, comparable to other studies (10, 12). The prevalence of patients with OPGs, who needed a treatment was 16.3% (14/86) in our cohort. Typically, OPGs are WHO grade I glial neoplasms, termed pilocytic astrocytomas, which require chemotherapy in 13-15% of patients (13).
NF1 patients are known to be predisposed to a wide spectrum of malignancies. These include myeloid disorders, such as juvenile chronic myelogenous leukemia (JCML), acute myeloid leukemia (AML), pheochromocytomas, duodenal carcinoid, hepatoblastoma, somatostatinoma and parathyroid adenoma. After MPNST, rhabdomyosarcoma (RMS) is the second most common type of non-neurogenic sarcoma in NF1 (10). Numerous of these neoplasms were observed in our patients with NF1. Also, in the group of patients with multiple malignancies, most of them (4 out of 6) were NF1-individuals.
Pheochromocytoma may occur in 0.1-7.7% of patients presenting with NF1 (14). The frequency of adrenal tumors in NF1-patients was 0.5% (4/843) in our cohort, and two of these tumors were pheochromocytomas. The pheochromocytoma is also part of the vHLS, with a prevalence of about 20%. vHLS disease predisposes an individual to the development of different types of tumor in bilateral and multicentric forms. Besides pheochromocytoma, retinal angiomas, hemangioblastomas of the central nervous system (mainly cerebellum and spinal cord), renal cysts, clear cell carcinomas, pancreatic cysts, neuroendocrine pancreatic tumors, tumors of the endolymphatic sac and epididymal cystadenoma may develop in various combinations in vHLS patients (15). We provided medical care for five patients with vHLS. Malignancy occurred in one of them, and this patient had three different neoplasms, and pheochromocytoma was not diagnosed. Mutations in VHL gene are most often associated with clear cell renal carcinoma, the most common type of renal cancer. They also play a major role in sporadic cases of clear cell renal carcinoma (16), which was diagnosed in our vHLS patient.
One of the patients with multiple malignancies was a NF2-individual that developed, two types of tumors: MPNST and brain tumor. In all the NF2-population, the prevalence of malignant tumors was 27.8% (5/18) in our cohort. This incidence was higher in comparison to other reports. The development of malignant tumors in NF2 is relatively rare in contrast to its counterpart NF1, and MPNSTs are unusual in NF2. Most MPNSTs present with a significant spindle cell content, although <5% of MPNSTs have a mixed epithelioid spindle composition, in a variant known as epithelioid malignant peripheral nerve sheath tumor (EMPNST) (17, 18). NF2 predisposes to the development of benign nerve sheath tumors that are predominantly schwannomas, meningiomas, and low-grade ependymomas (19).
TSC is generally characterized by the presence of benign tumors, but some patients with malignancies have been reported in the literature. The prevalence of malignant tumors was 8.3% (3/36) in our cohort of TSC patients, including two brain tumors and one renal cancer. Peron et al. examined a large Italian TSC population (240 patients aged from 3 months – 74 years), assessing the frequency of malignancies to determine whether there is an increased risk for cancer in this disorder, and looking for possible features associated with the development of neoplasia. Fifteen patients had malignancies (6.25%); five of seven renal tumors were renal cell carcinomas. Eight patients had a non-renal tumor (3.3%), and no patients developed more than one malignancies. The prevalence of malignant tumors was comparable with the prevalence in the general population (5.6% vs. 4.4% in Italy). Median age at cancer diagnosis was lower (37.5 vs. 66.0 years). This study demonstrated also that TSC patients do not seem to have an increased risk for malignancies besides renal cell carcinoma. However, when cancer develops, age at diagnosis is lower than that in the general population (20).
In conclusion, the prevalence of malignancies in our large cohort of children and adults with neurocutaneous disorders (phacomatoses) was 9.4%, 9.1% in patients with NF1, 27.8% in NF2 and 8.3% in TSC. In comparison to the general population, the risk of malignancy in patients with phacomatoses was 21.3-fold higher. Finally, multiple malignancies were diagnosed in 0.7% of all patients with phacomatoses and 7% among those patients already diagnosed with a malignancy.
- Received June 28, 2018.
- Revision received July 13, 2018.
- Accepted July 16, 2018.
- Copyright© 2018, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved





{kind=link}
{kind=link}