


Abstract
Background/Aim: Organ transplant patients treated with the immunosuppressive drug cyclosporine A often present malignant kidney tumors. Cyclosporine A can promote oncogenesis in a cell-intrinsic manner by increasing the production of vascular endothelial growth factor (VEGF). Materials and Methods: We explored the impact of cyclosporine A and the role of the unfolded protein response (UPR) on three human renal cell carcinoma (RCC) cell lines under normoxic and hypoxic (1% O2) conditions. Results: Cyclosporine A regulated the expression of VEGF at the post-transcriptional level. Cyclosporine A induced the inositol requiring enzyme-1α (IRE1α) arm of the UPR and stabilized neosynthesized proteins in RCC cells. Toyocamycin, an inhibitor of IRE1α, abolished the clonogenic growth of RCC cells and reduced induction of VEGF by cyclosporine A under hypoxia. Conclusion: Our findings highlight the impact of cyclosporine A on the proteostasis of RCC cells, and suggest the potential therapeutic interest of targeting the UPR against tumors arising in the context of organ transplantation.
- Cyclosporine A
- renal cell carcinoma
- unfolded protein response
- vascular endothelial growth factor
- toyocamycin
Cyclosporine A is one of the most frequently used immunosuppressive drugs following organ transplantation. Cyclosporine A prevents acute rejection of the transplanted organ by its recipient. However, long-term complications, including various types of malignancy, are emerging as a factor limiting patient survival (1, 2). Malignant tumors, in particular those originating from the kidney, occur with a five-fold increased frequency in organ transplant patients (1). The mechanisms that promote the appearance of malignant kidney tumors, in particular renal cell carcinoma (RCC), under immunosuppressive treatment are complex and probably multifactorial. Several studies indicate that cyclosporine A promotes pro-tumorigenic signaling in a cell-intrinsic manner in RCC cells (3, 4). Vascular endothelial growth factor (VEGF) produced by RCC cells is a possible actor in cyclosporine A-induced carcinogenesis, since cyclosporine A increases its expression in RCC cells (5). VEGF is usually regarded as an essential growth factor in RCC, considering that these tumors are highly vascular and that multiple alterations of angiogenic signaling are found in RCC (6-8). In addition to controling tumor angiogenesis, VEGF is implicated in autocrine/paracrine signaling in RCC cells (9, 10). The clinical efficacy of sunitinib, an inhibitor directed against the VEGF receptor, constitutes further proof that the VEGF receptor is an essential actor in renal carcinogenesis (11).
While VEGF is likely an important factor and a target for the treatment of RCC, it is still unclear how RCC cells regulate the production of VEGF upon exposure to cyclosporine A. Early studies suggested the possible importance of transcription in the regulation of VEGF expression (5). These findings were challenged by other studies which reported that post-transcriptional mechanisms account for most of the regulation of VEGF production (12, 13). Recently, cyclosporine A was also found to interfere with the functionality of the secretary compartment of eukaryotic cells (14, 15). For example, by interacting with cyclophilin B, a member of the cyclophilin family of proteins present in the endoplasmic reticulum (ER) of eukaryotic cells, cyclosporine A interferes with the folding and the turn-over of secreted proteins (14, 15). The possibility that cyclosporine A interferes with cellular proteostasis, i.e. the biogenesis, folding, trafficking and degradation of proteins within eukaryotic cell, has not yet directly been addressed in RCC cells.
The unfolded protein response (UPR) is a homeostatic mechanism that regulates the functionality of the secretion apparatus of eukaryotic cells (16-18). The UPR consists of three transduction cascades that are activated under conditions of ER stress, and that are each regulated by a distinct protein sensor: i) Protein kinase R-like endoplasmic reticulum kinase (PERK) is able to phosphorylate the eukaryotic translation initiation factor-2α (eIF2α) and control global protein synthesis; ii) Inositol-requiring enzyme-1α (IRE1α) is a nuclease reported to catalyze the unconventional splicing of the mRNA encoding the X-box binding protein-1 (XBP1) to produce a splice variant of this transcription factor, sXBP1. iii) Activating transcription factor-6 (ATF6) regulates the expression of specific genes. Interestingly, the activated IRE-1α and PERK arms of the UPR were found to exert a positive regulatory effect on the expression of VEGF in multiple experimental systems, although to our knowledge no study was devoted to RCC (13, 19-22). In the present study, we aimed to address the potential impact of cyclosporine A on the proteostasis of RCC cells and to test whether the UPR represents a valid therapeutic target in RCC.
Materials and Methods
Cell culture and reagents. The three human RCC cell lines used in this study were ACHN (ATCC-CRL-1611), Caki-1 (ATCC-HTB-46) and 786-O (ATCC-CRL-1932), and were purchased from American Type Culture Collection (LGC standards, Strasbourg, France). Hypoxic conditions were obtained by placing cells in a hypoxic chamber with 1% O2 (Sanyo, Roissy, France). GSK2606414, puromycin and tunicamycin were purchased from Calbiochem. Tacrolimus and all other reagents were purchased from Sigma (St Quentin Fallavier, France).
Determination of VEGF levels. VEGF and amphiregulin (AREG) concentrations in the cell culture medium were measured with an ELISA kit purchased from R&D systems (Lille, France) (for VEGF, limit of detection <5 pg/ml, limit of quantification <15.6 pg/ml; for AREG, limit of detection <1.4 pg/ml, limit of quantification <15.6 pg/ml). Both kits were used according to the manufacturer's instructions.
Quantitative Polymerase Chain Reaction (PCR). Total RNA was extracted using RN-Easy Mini Kit (Qiagen, Courtaboeuf, France) and reverse-transcribed using High Capacity cDNA Reverse Transcription kit and random hexamer (Applied Biosystems, Courtaboeuf, France). Amplification was performed with the TaqMan Universal PCR master Mix (Applied Biosystems) on an ABI 7900HT Sequence Detection System (Applied Biosystems) using primers and probe sets for VEGFA and glyceraldehyde-phosphate dehydrogenase (GAPDH) (TaqMan Gene Expression Assay; Applied Biosystems).
Determination of VEGFA promoter activity. The DNA construct covering the human VEGFA promoter inserted in the vector pGL3, encoding firefly (Photinus pyralis) luciferase (Promega, Charbonnières-les-Bains, France) was previously reported by Miyagi et al. (23). Cells were transfected using the Genjet reagent, as indicated by the provider (Tebu-Bio, Le Perray en Yvelines, France). Lysis was performed using appropriate reagents from Promega, and luciferase activity was measured on a CentroLB 960 Microplate Luminometer (Berthold, Thoiry, France).
RNA interference. Small interfering RNA (siRNA) directed against XBP1 (references S14915 and S14913, both from Applied Biosystems) were transfected using the siPORT-NeoFX reagent (Invitrogen, Courtaboeuf, France) and Opti-MEM transfection medium (Life Technologies, Courtaboeuf, France), according to the manufacturer's instructions.
Labelling of neosynthesized proteins with low doses of puromycin. The incorporation of puromycin into nascent proteins and its subsequent detection with a monoclonal antibody directed against puromycin were previously reported by Schmidt et al. (24). Briefly, cells were exposed to 10 μg/ml puromycin for 10 min. Cycloheximide, an antibiotic that blocks protein synthesis in eukaryotic cells, was used as a negative control and applied 30 min before exposure to puromycin at a concentration of 50 μM. Puromycinylated proteins were detected by immunoblot analysis.
Immunoblotting. Complete cell extracts were transferred to nitrocellulose membranes using standard procedures. The ECL reaction was used to reveal protein. Rabbit antibodies directed against sXBP1, glucose-regulated protein-78 (GRP78), eIF2α and its phosphorylated form (p-eIF2α) were from Cell Signaling (Danvers, MA, USA). Mouse anti-puromycin (MABE 343) was purchased from Merk Millipore (Saint Quentin en Yvelines, France). Rabbit anti-oxygen-regulated protein-150 (ORP150) was from Abcam (Cambridge, UK). Mouse anti-β-actin was obtained from Sigma. Secondary antibodies coupled to horseradish peroxidase were purchased from GE Healthcare.
Clonogenicity assay. Clonogenic growth was measured as previously described (25). Briefly, 200 viable RCC cells were seeded per well of 15 mm diameter. Cells were allowed to adhere overnight, and later exposed to cyclosporine A. After 10 days of culture, the clones were stained with Giemsa and a blind count was performed.
Statistical analyses. Student's t-test was used for comparison between groups and a value of p<0.05 was considered as threshold for significance. Immunoblots were scanned and quantified using the software Image J (National Institute of Health, USA).

Cyclosporine A (CsA) increases vascular endothelial growth factor (VEGF) production by renal carcinoma cells (RCC) under hypoxic conditions. The indicated RCC cell lines were exposed to cyclosporine A (1 and 5 μM) and tacrolimus (Tac; 1 μM) under normoxic and hypoxic conditions (1% O2). VEGF production was determined in cell culture supernatants after 18 h at these conditions. Fold induction of VEGF concentrations measured in hypoxic over normoxic conditions was calculated. *Statistically significantly different at p<0.05 compared to the control. The results are from a representative experiment performed in triplicate.
Results
Cyclosporine A increases VEGF production by RCC cells under hypoxic conditions. In order to examine the impact of cyclosporine A on RCC cells, we exposed three RCC cell lines with different genetic backgrounds to cyclosporine A (1 and 5 μM) and tacrolimus (1 μM) in vitro. Cells were maintained for 18 h in normoxic and hypoxic conditions (1% O2) and cell culture supernatants were collected and analyzed by ELISA to determine VEGF production (Figure 1). Hypoxic induction of VEGF was found in all three RCC cell lines, although the induction was more pronounced in Caki-1 cells. Cyclosporine A increased the hypoxic induction of VEGF, while no induction of VEGF was apparent with tacrolimus (Figure 1). Under our conditions, cyclosporine A did not modulate the activation levels of the main oncogenic kinase cascades: protein kinase B (PKB)-mammalian target of rapamycin (mTOR) or extracellular signal-regulated kinases (ERK) (data not shown). The transcription factor hypoxia-inducible factor-2α (HIF2α), the main inducible isoform of HIF that we detected under our experimental conditions, was induced to the same extent in RCC cells irrespective of their exposure to cyclosporine A (data not shown). We analyzed the VEGFA mRNA levels and the activity of the corresponding promoter in Caki-1 cells (Figure 2). Hypoxic conditions induced a clear increase in the expression of VEGFA mRNA, but cyclosporine A had no clear effect on this parameter (Figure 2A). The promoter activity of VEGFA was also studied with a construct encompassing the 6-kb region located upstream of the translation initiation site (Figure 2B). The activity of the VEGFA promoter was induced by more than 15-fold by hypoxia, but cyclosporine A exerted no further stimulation on its promoter activity (Figure 2B). These findings indicate that cyclosporine A mostly influenced the production of VEGF through post-transcriptional mechanisms in our experimental setting.
Cyclosporine A activates the IRE1α arm of the UPR in RCC cells. In order to explore the possible regulatory role of the UPR in the transcriptional regulation of VEGF expression, we examined the activity of the three arms of the UPR (IRE-1α, PERK and ATF6) in Caki-1 cells exposed to cyclosporine A (Figure 3A). We monitored the appearance of the spliced form of XBP1 as a marker of IRE1α activation, the phosphorylation status of the eIF2α on Ser51, which reflects the activity of the kinase PERK, and the levels of the chaperones GRP78 and ORP150, whose expression is under transcriptional control by ATF6 (26). We observed that cyclosporine A induced the activation of the IRE-1α arm of the UPR, yet to a lesser extent under hypoxic than normoxic conditions (Figure 3A). We decided to explore the potential significance of sXBP1 because this transcription factor had previously been shown to regulate the transcriptome of cancer cells under hypoxic conditions (22). Two independent siRNAs targeting XBP1 were used, which prevented the appearance of sXBP1 upon tunicamycin exposure (Figure 3B). Neither had a detectable effect on the VEGFA mRNA level in Caki-1 cells exposed to hypoxic conditions and cyclosporine A (Figure 3C), suggesting that the UPR is not an essential regulator of VEGFA transcription in this situation.

Cyclosporine A (CsA) predominantly regulates vascular endothelial growth factor A (VEGFA) expression post-transcriptionally. A: Quantitative analysis of VEGFA mRNA level in Caki-1 cells. The VEGFA mRNA level was analyzed in Caki-1 cells exposed to cyclosporine A (1 μM) under normoxic and hypoxic conditions (1% O2) for 18 h. Results are from an experiment performed in triplicate. The VEGFA mRNA levels were normalized to those of glyceraldehyde-phosphate dehydrogenase (GAPDH), taking the control condition (Ctl) as reference. B: Luciferase activity for the analysis of the promoter of VEGFA. A construct encompassing the 6-kbp region located upstream of the translation initiation site was transfected into Caki-1 cells, exposed to cyclosporine A in normoxic and hypoxic conditions for 18 h. The upper panel contains a graphical description of the VEGFA promoter, including the localization of the binding sites for the following important transcription factors: XBP1: X-box binding protein-1; AP1: Activator Protein 1; HIF1: Hypoxia-Inducible Factor; SP1 and SP3 transcription factors. N.S: Lack of significant difference compared to cells under the same conditions treated with cyclosporine A.
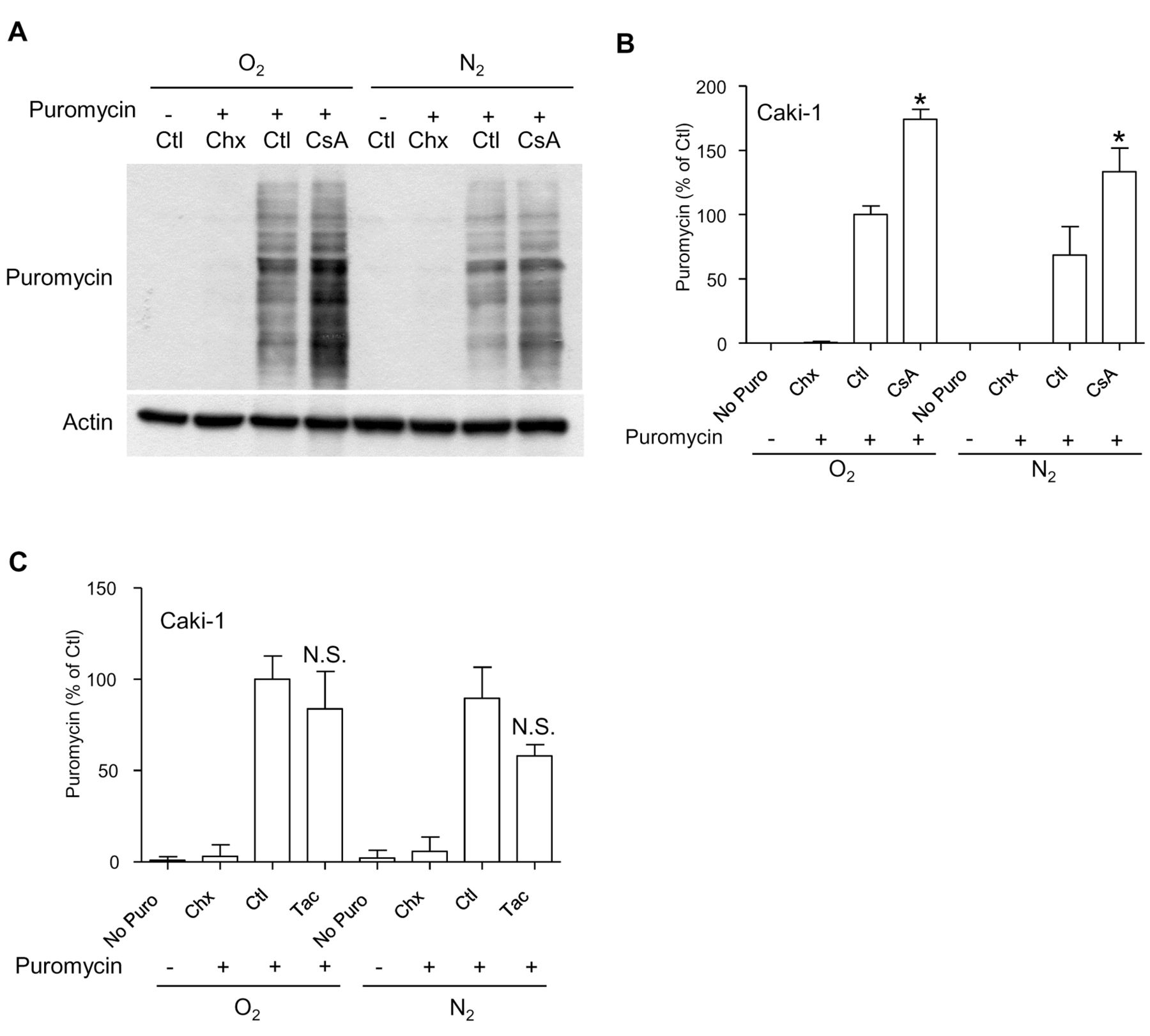
Cyclosporine A alters the nascent proteome of RCC cells. In an attempt to examine the effect of cyclosporine A on the secretion of proteins other than VEGF, we tested the effect of cyclosporine A on the production of other growth factors produced in the culture supernatants of RCC cells. Interestingly, cyclosporine A increased the amount of AREG produced by Caki-1 cells under hypoxic conditions in a manner that was reminiscent of the effect observed with VEGF (data not shown). This suggests that cyclosporine A exerts a general effect on tumor cell proteostasis rather than a specific regulatory effect on the transcription of VEGF. In order to explore the regulation of the nascent proteome in RCC cells, we used a technique that relies on the incorporation of low concentrations of puromycin into nascent proteins (24). Cells exposed to different culture conditions were labelled with puromycin for a short interval of time. Cellular extracts were prepared and puromycinylated proteins were analyzed by immunoblotting (24). Interestingly, cyclosporine A increased the overall levels of puromycinylated proteins by almost two-fold in RCC cells under normoxic and hypoxic conditions, revealing a broad effect of cyclosporine A on the nascent proteome of RCC cells (Figure 4A and B). This effect was found in all RCC cell lines tested (data not shown), and was also specific to cyclosporine A, since tacrolimus did not show such activity (Figure 4C). We noticed an apparent inverse correlation between the level of puromycinylated proteins and the induction of the sXBP1 previously observed (Figure 3). This observation suggests the possibility that the effect of cyclosporine A on the nascent proteome is a possible explanation for the induction of the UPR.
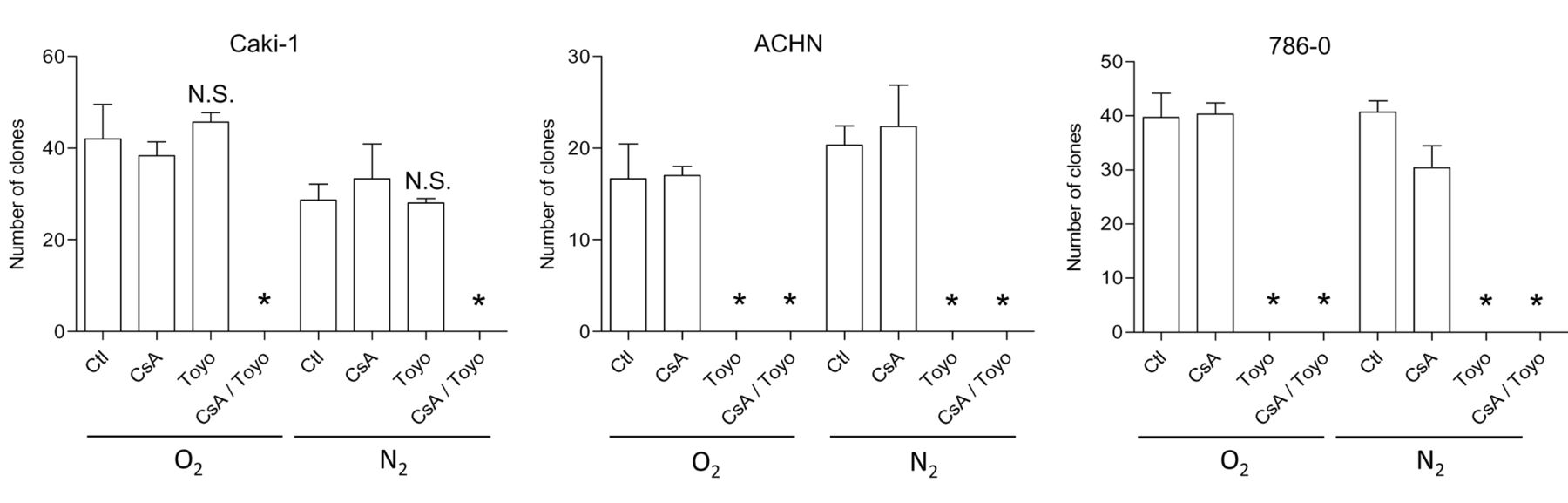
Targeting the IRE1α arm of the UPR in RCC cells. Based on the observations that cyclosporine A activates the IRE1α arm of the UPR, we decided to explore the effects of two chemical inhibitors of the UPR on RCC cells, namely toyocamycin, a compound produced by some Actinomycete strains and recently reported to inhibit IRE-1α-dependent RNAse activity (27, 28); and GSK2606414, a chemical blocker of the kinase PERK (29). Both inhibitors were applied at a concentration of 1 μM, shown to inhibit their respective targets (Figure 5A and B) without exerting a direct short-term cytotoxic effect as single agents (data not shown). While GSK2606414 displayed no significant effect on the amount of VEGF produced in the cell culture medium of Caki-1 cells, toyocamycin dramatically reduced the amount of VEGF produced in hypoxic cells exposed to cyclosporine A (Figure 5C). We explored the anti-oncogenic efficacy of toyocamycin with a clonogenic growth assay (Figure 6). A combination of cyclosporine A and toyocamycin radically prevented the clonogenic growth of Caki-1 cells. The clonogenic growth of 786-O and ACHN cells was inhibited upon the application of toyocamycin as a single agent (Figure 6). The findings suggest that the IRE1α arm of the UPR may be a potential therapeutic target in RCC cells, especially upon exposure to cyclosporine A.

Cyclosporine A (CsA) activates the inositol requiring enzyme-1α (IRE1α) arm of the unfolded protein response in renal cancer cells. A: Protein extracts were prepared from Caki-1 cells exposed to cyclosporine A for the indicated time under normoxic and hypoxic conditions, and various UPR markers were analyzed by immunoblotting. Tunicamycin was used as an inducer of endoplasmic reticulum stress and was applied at a concentration of 10 μM for 6 h. B. Caki-1 cells were transfected with two siRNAs targeting distinct regions of X-box binding protein-1 (XBP1), and exposed to tunicamycin (10 μM) for 6 h. An immunoblot analysis was subsequently performed using an antibody to XBP1, revealing a more than 95% inhibition of the knock-down. C: Caki-1 cells were treated as indicated in panel B and exposed to cyclosporine A (1 μM) under normoxic and hypoxic conditions for 18 h. The level of VEGFA mRNA was measured and normalized to glyceraldehyde-phosphate dehydrogenase (GAPDH), taking the control condition (Ctl) as reference. N.S.: Lack of significant difference compared to the same conditions with control siRNA.
Discussion
Cyclosporine A, a calcineurin inhibitor that is widely used to prevent organ rejection, increases the production of VEGF and sustains malignant transformation of RCC cells in a cell-intrinsic manner (3, 4). We explored the impact of cyclosporine A on RCC cells with a focus on the UPR and the regulation of VEGF. We observed that cyclosporine A essentially activates the IRE1α arm of the UPR, and explored the consequences of its pharmacological targeting by applying the inhibitor toyocamycin (27, 28). In Caki-1 cells, toyocamycin prevented the boost in VEGF production induced by cyclosporine A under hypoxic conditions. In the RCC cell lines tested here, toyocamycin exerted a strong anti-oncogenic activity, both as a single agent and in combination with cyclosporine A. The present study, therefore, highlights the broad impact of cyclosporine A on the proteostasis of RCC cells and suggests, as far as we are aware, for the first time that the UPR might be a potentially interesting target in therapy against these tumors.

Cyclosporine A, but not tacrolimus, alters the nascent proteome of renal carcinoma cells. A: Caki-1 cells were exposed to cyclosporine A (1 μM) under normoxic and hypoxic conditions (1% O2) for 18 h, and nascent proteins were labelled with puromycin (10 μg/ml) for 10 minutes. Where indicated, cells were pre-incubated for 30 min with cycloheximide (Chx, 50 μM) in order to block protein translation. We present a single representative immunoblot experiment where puromycinylated proteins were detected. B: Quantification of blots based on three independent experiments, with the control condition (Ctl) taken as the reference value. *Significantly different at p<0.05 compared to the same conditions without cyclosporine A. C: Quantification based on data obtained from Caki-1 cells exposed to tacrolimus (1 μM) under normoxic and hypoxic conditions (1% O2) for 18 h. N.S.: Lack of a significant difference compared to the same conditions without tacrolimus.
The present study aimed to explore the possibility that RCC cells might differ in their response to cyclosporine A. The three RCC cell lines used in this study were chosen because their mutational status differs at the level of three essential oncogenes reported to play a role in renal carcinogenesis (the genes TP53, PTEN and VHL). Interestingly, we found that the effect of cyclosporine A on VEGF was relatively independent of the genotype of the RCC cell line examined. Cyclosporine A increased the secretion of VEGF and exerted comparable effect on the nascent proteome in all three RCC cells. The present findings do not rule-out the possible contribution of individual genomic polymorphisms to the control of VEGF production in RCC cells. Nevertheless, the observation that cyclosporine A had an effect on RCC cells with different genomic backgrounds is in agreement with the presumed mode of action of cyclosporine A on tumor proteostasis, and with the notion that cyclosporine A targets conserved actors of proteostasis, such as cyclophilin B in the secretory compartment (14). More studies are required to examine the regulation of the UPR in different RCC cells, but the UPR is emerging as a central regulator of human tumorigenesis (16). Core actors of the UPR, such as IRE1α, might constitute functionally important and relatively well-conserved targets among tumors arising in a context of proteostasis disturbance induced by chronic exposure to cyclosporine A.
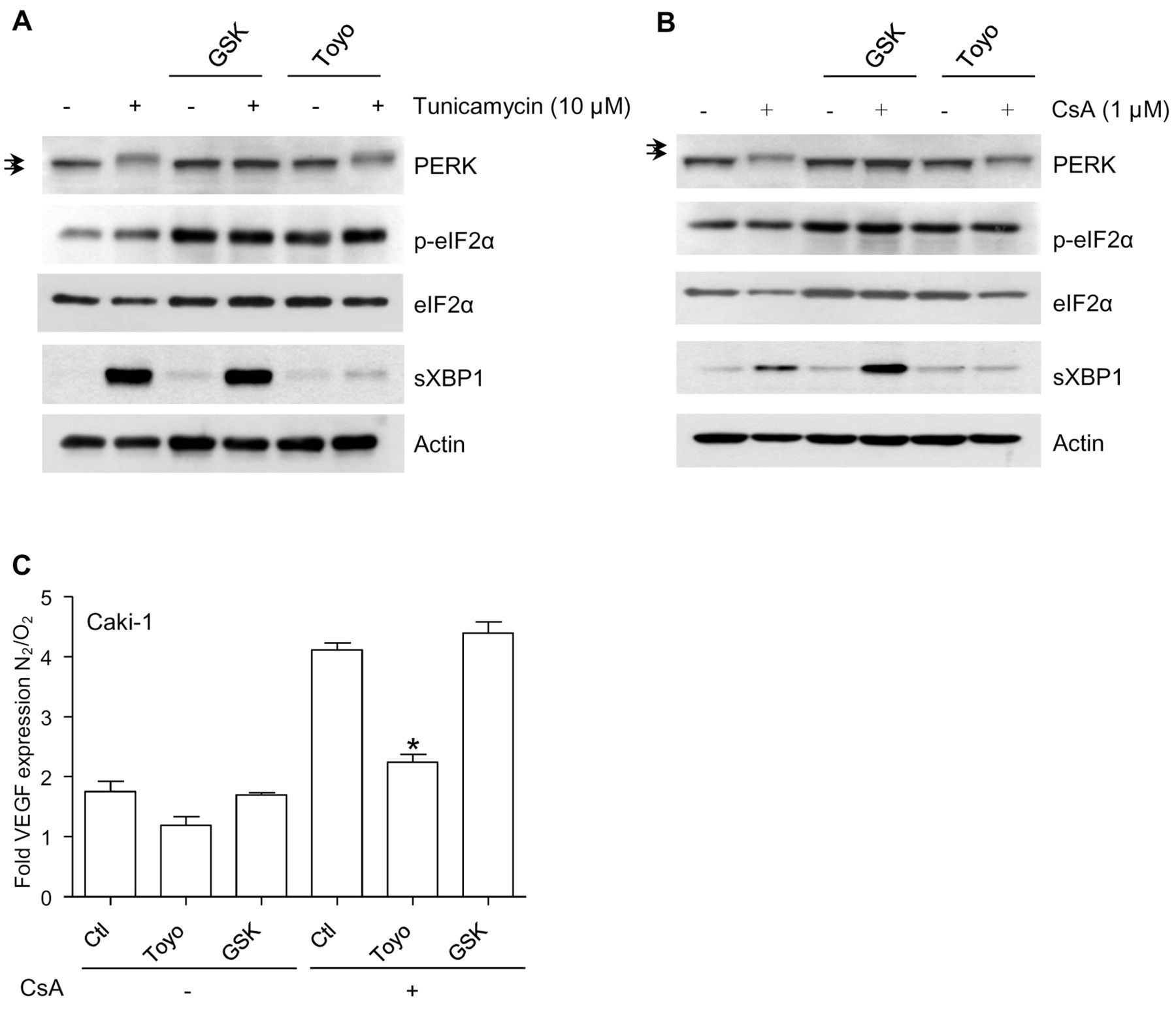
In vitro study of the effects of chemical inhibitors of the unfolded protein response in renal carcinoma cells exposed to cyclosporine A (CsA). A: The chemical inhibitors toyocamycin (Toyo; 1 μM) and GSK2606414 (GSK; 1 μM), directed against inositol requiring enzyme-1α (IRE1α) RNase activity and the protein kinase R-like ER kinase (PERK), respectively, were applied 30 min before tunicamycin (10 μM, 6 h) and the indicated markers were analyzed by immunoblot. The arrows indicate the existence of a mobility shift applying to PERK in conditions of endoplasmic reticulum stress and prevented by GSK2606414. B: Toyocamycin (1 μM) and GSK2606414 (1 μM) were applied 30 min before cyclosporine A (1 μM, 6 h) and the indicated markers were analyzed by immunoblot. C: Caki-1 cells were exposed to cyclosporine A (1 μM), toyocamycin (1 μM) and GSK2606414 (1 μM), applied as indicated under normoxic or hypoxic conditions (1% O2), and vascular endothelial growth factor (VEGF) production was determined in cell culture supernatants after 18 h. Results are presented as a fold induction of VEGF measured under hypoxic over normoxic conditions. *Significantly different at p<0.05 compared to equivalent conditions without toyocamycin or GSK2606414.
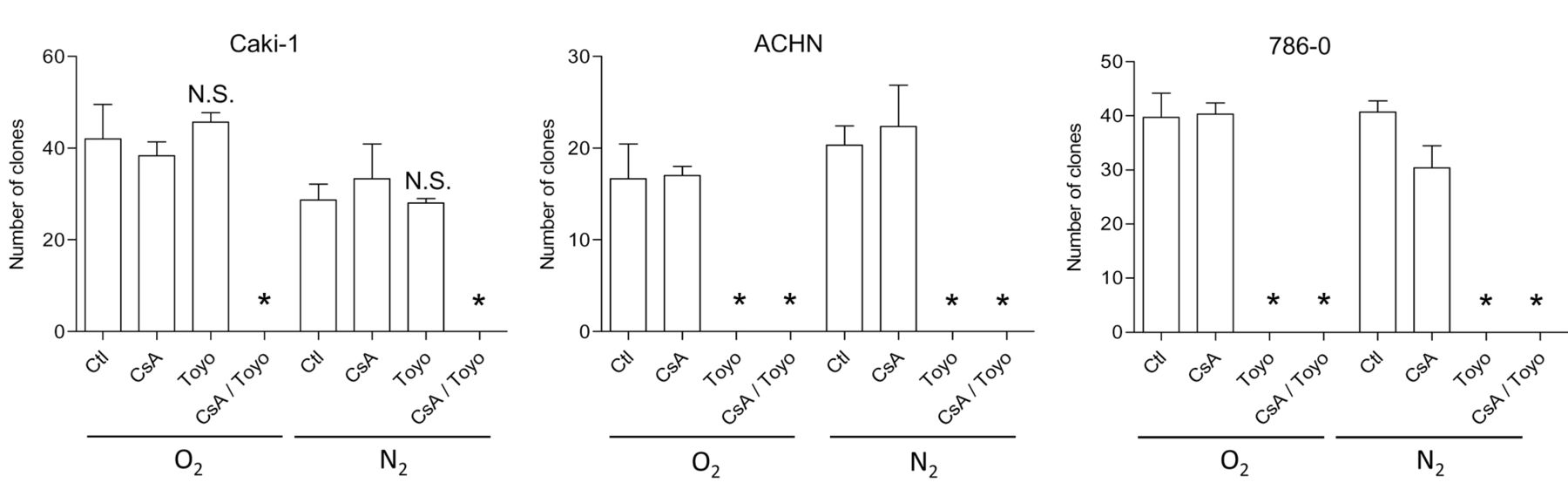
Clonogenic growth of renal carcinoma cells exposed to cyclosporine A (CsA) and toyocamycin (Toyo). The clonogenic growth of each RCC cell line was measured upon exposure to cyclosporine A (1 μM) and toyocamycin (1μM), applied as indicated under normoxic or hypoxic conditions (1% O2). The results are from a representative experiment performed in triplicate. Ctl: Control. *Significantly different at p<0.05 compared to conditions with CsA alone.
Additional studies are required to explore the role and the regulation of the UPR in RCC occurring after organ transplantation. In this respect, an obvious limitation of our study is its experimental set up based on the in vitro exposure of RCC cells to cyclosporine A. In order to explore the potential of the UPR as a therapeutic target in cyclosporine A-associated carcinogenesis in a more relevant manner, future investigations should be centered on human tumor samples. These studies should explore the impact of cyclosporine A on the proteostasis of cancer cells, with a focus on ER chaperones and the IRE1α arm of the UPR. Based on our in vitro results, we propose that the study of tumor proteostasis offers promising medical perspectives for post-transplant RCC.
Acknowledgements
The Authors thank Dr. Zuzana Saidak for helpful comments on the draft. We are grateful for financial support received from Association Santelys, Ligue contre le Cancer, comité de la Somme, and CHU Amiens.
Footnotes
Conflicts of Interest
The Authors have not declared any conflict of interest.
- Received January 3, 2017.
- Revision received February 7, 2017.
- Accepted February 14, 2017.
- Copyright© 2017, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}