


Abstract
Background: Protein tyrosine kinases (PTKs) play major roles in signal transduction during cell proliferation and apoptosis. Tyrphostin AG17 was previously shown to be a potent tumor growth inhibitor, while AG17 induced apoptosis and inhibited activity of cyclin-dependent kinase 2. We herein describe the binding features of tyrphostin AG17 analogs, such as TX-1123, with Src kinase (Src-K). Materials and Methods: Structural data for Src-K were obtained from a protein data bank (ID=2SRC), and the molecular interactions between Src-K and TX-1123 derivatives were examined. Results: TX-1123 exihibited potent Src-K inhibitory activity (half maximal-inhibitory concentration=2.2 μM), and fit into the pocket of the Src-K molecule as well as c-AMP did. Conclusion: The binding profiles of TX-1123 derivatives differed from each other, while their Src-K inhibitory activities were affected by their fit in the Src-K molecule.
Protein tyrosine kinases (PTKs) are members of a large family of oncoproteins and proto-oncoproteins that play major roles in signal transduction during normal cell division, terminal cell differentiation, and apoptosis (1-5). PTK activity is associated with proliferative disorders such as cancer, and PTK inhibitors were developed as potential therapeutic agents (6-8). Tyrphostins were examined as low-molecular-weight PTK inhibitors designed to compete with substrate rather than ATP (9-10).
We previously reported that tyrphostin AG17 suppressed the effects of insulin in rat white adipocytes (11). Palumbo et al. demonstrated that AG17 induced apoptosis and inhibited cdk2 activity through a mechanism that purportedly does not involve reductions in cellular ATP level (12). Based on the potential mechanisms underlying the antiproliferative effects of AG17, it is important to note that AG17 (as SF6847, an alternate name for AG17) has been shown to act as an uncoupler of oxidative phosphorylation in isolated rat liver and heart mitochondria (13). We previously described a new tyrphostin analog of AG17 that has lower mitochondrial toxicity, TX-1123 (14). In the present study, we analyzed the kinase-inhibitory activities of TX-1123 derivatives (Figure 1), and examined the binding profiles of these TX-1123 family members using docking simulation.
Materials and Methods
Inhibition of Src kinase activity. PTK activity was summarized from our previous report (14). From the enzyme activity (as a percentage to that of the control) as a function of drug concentration, the half maximal-inhibitory concentration (IC50) value was estimated as the index of the enzyme inhibition of the designed compound.
Analysis of interaction of TX-1123 derivatives with kinase. Src kinase structure was obtained from a protein data bank (PDB ID=2SRC). The interactive analysis between TX-1123 derivatives and Src kinase was examined by the molecular simulation technique (MM-MD) using insightII discover under consistent valence force field (Accelrys Inc., San Diego, CA, USA) and molegro virtual docker (CLC bio, Aarhus, Denmark), as previously described (15, 16).
Results
Inhibition of Src kinase activity by TX-1123 derivatives. TX-1123 exhibited the most potent inhibition of Src kinase activity among the compounds tested, with an IC50 of 2.2 μM (Table I). Src kinase inhibition was suppressed by the addition of a methoxy group to TX-1123, and the IC50 of the resulting derivative TX-1925 was 3.1 μM. The replacement of a sterically-bulky t-butyl group in TX-1123 by methyl reduced its inhibitory activity, with an IC50 of the resulting derivative TX-1918 was 4.4 μM. The addition of a benzene ring to the cyclopentenedione site also reduced the inhibitory activity (TX-1926, IC50 >27 μM). The general mitochondrial uncoupling drug tyrphostin AG17 exhibited weak inhibition of Src kinase activity (IC50 >350 μM). Methoxy tyrphostin AG17 (TX-1927) had a similarly high IC50 >340 μM.

Structure of TX-1123 derivatives.
Ligand-binding pockets in Src kinase molecule. The Src kinase (protein data bank ID=2SRC) molecule flexibly moved during the MM-MD simulation period (data not shown). Three major ligand-binding pockets were detected in the Src kinase molecule (Figure 2A-C), with the main binding pocket (i.e. pocket A) being observed at the center of this molecule. Binding pocket B was identified based on the X-ray data of the ligand (c-AMP)-binding site with Src kinase (Figure 3A). Binding of ligand (e.g. TX-1123 derivatives) was not detected at pocket A or C in the present ligand-binding simulation.
Binding features of TX-1123 derivatives. TX-1123 bound at pocket B, and the binding position was shifted more to the right than that of c-AMP (Figure 3B). An interaction was observed between the phenolic hydroxyl group of TX-1123 and the phenolic hydroxyl group of Tyr416 in the Src kinase molecule (Figure 3C), and the total interaction energy was −115.808 kcal/mol (Table I). TX-1925 and TX-1918 exhibited Src kinase inhibition similarly to TX-1123 (Table I), and bound at pocket B, like TX-1123. The binding position of TX-1925 to Src kinase was shifted downward from that of c-AMP (Figure 4A), and the total interaction energy was −106.957 kcal/mol. The binding position of TX-1918 was also shifted downward from that of c-AMP (Figure 4B) and TX-1925, and the total interaction energy was −102.254 kcal/mol. TX-1918 interacted with the phenolic hydroxyl group of Phe405 in the Src kinase molecule through its own phenolic hydroxyl group (data not shown).
Half-maximal inhibitory concentration (IC50) for inhibition of protein Src kinase and interaction energy of TX-1123 derivatives.
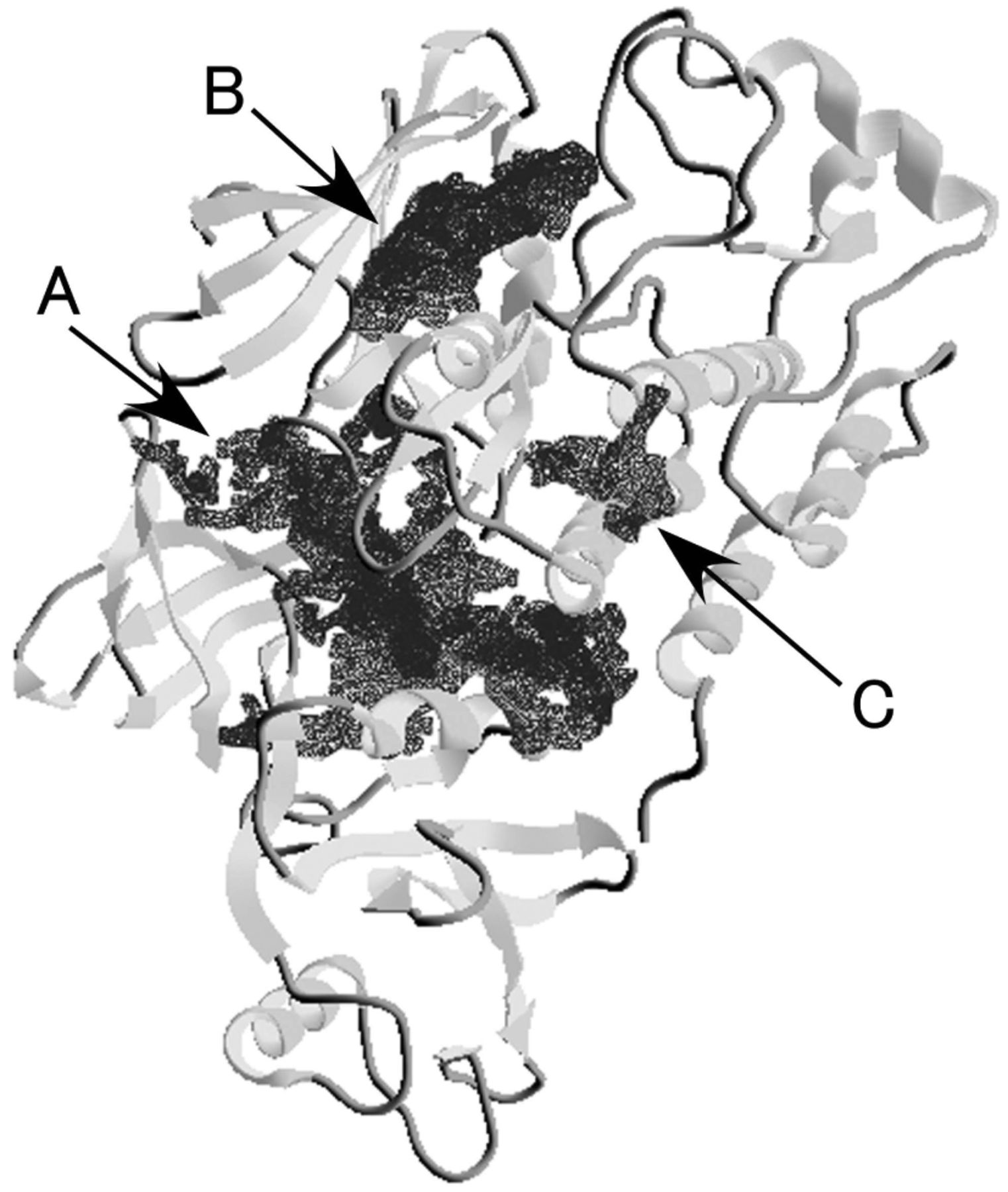
Binding pockets of the Src kinase molecule. Ligand-binding pockets (A-C) were determined by molecular simulation.
The binding position of TX-1926 was near to that of c-AMP (Figure 4C). TX-1926 showed good binding energy (−116.078 kcal/mol) similar to that for TX-1123, whereas its kinase inhibitory activity was weaker than those of TX-1123, TX-1925, and TX-1918. The binding position of TX-1927 was almost the same as that of TX-1123 (Figure 4D). TX-1927 interacted with the phenolic hydroxyl group of Tyr416 and amino group of Gly276 of Src kinase through its own phenolic hydroxyl group and cyano group, respectively (data not shown). The binding energy and Src kinase inhibition of TX-1927 (−98.813 kcal/mol, IC50 >340 μM) were significantly weaker than those of TX-1123. AG17 bound at different sites in Src kinase to the TX-1123 derivatives, and the phenolic hydroxyl group of Tyr90 interacted with cyano group of AG17. The two carboxyl groups of Tyr149 and Gln144 of Src kinase interacted with the phenolic hydroxyl group of AG17 (Figure 4F). The binding energy of AG17 (−98.821 kcal/mol) was equivalent to that of TX-1927.

Binding profile of ligands to Src kinase. A: c-AMP fitted in pocket B (see Figure 2) of Src kinase. B: TX-1123 fitted in pocket B as well as c-AMP did. C: The phenolic hydroxy group of TX-1123 interacted with Tyr416 of Src kinase.

Interaction profiles of TX-1123 derivatives with Src kinase. TX-1925 (A), TX-1918 (B), TX-1926 (C), and TX-1927 (D) fitted in Src kinase pocket B. Tyrphostin AG17 fitted in a different Src kinase site (pocket A) from TX-1123 derivatives. The cyano group of tyrphostin AG17 interacted with the hydroxyl group of Src kinase Tyr90, and the phenolic hydroxyl group of tyrphostin AG17 interacted with two carboxyl groups of Src kinase Tyr149 and Gln144 (F).
Discussion
2-Arylidine-4-cyclopentene-1,3-diones, which are unique soft acid-type electrophilic cyclopentene-1,3-dione moieties, were reported to be antitumor compounds (17). We previously developed non-nitro radiosensitizing hypoxic cytotoxin 2-hydroxyarylidene-4-cyclopentene-1,3-diones such as KIH-200, KIH-201, and KIH-202 (18). These synthesized KIH family members were found to be potent inhibitors of phosphate transport in mitochondria (19). We also developed the hydroxy-benzylidene-cyclopentenedione compound as a phenolic enhancer of chemiluminescence and found that KIH-201 was a potent enhancer of the luminol-hydrogen-horseradish peroxidase peroxide reaction (20). These cyclopentenedione moieties instead of the malononitrile moieties found in tyrphostin AG17 were expected to result in reduced mitochondrial toxicity. Tyrphostin AG17 exhibited potent mitochondrial cytotoxicity, such as mitochondrial uncoupling activity (Cmax=0.02 μM, Vmax=270 nanoatom O/mg/min) and inhibition of ATP synthesis (IC50=0.0035 μM) (14). TX-1123 was a designed tyrphostin AG17 analog, and exhibited lower mitochondrial toxicity [uncoupling activity (Cmax=2 μM, Vmax=260 nanoatom O/mg/min) and inhibition of ATP synthesis (IC50=5 μM)].
Different binding characteristics of TX-1123 family members to the Src kinase molecule compared with those of c-AMP binding were observed in a binding simulation. In general, native protein kinase was found to exist with a solvent molecule (e.g. water), and some solvent molecules were included in the kinase pocket (21). The X-ray data for c-AMP interaction with Src kinase is from one moment in time, and reflects neither molecular fluctuation nor interaction with solvent (22-25). A molecular docking simulation is considered appropriate because the verified interactive analysis between a ligand (e.g. TX-1123 family member) and protein (e.g. Src kinase) is performed with various solvents.
TX-1123 bound to a site near the c-AMP-binding pocket in the Src kinase molecule, and inhibited kinase activity. The phenolic hydroxy group of TX-1123 interacted with Src kinase Tyr460, and the methylation of this hydroxy group, such as in TX-1925, inhibited kinase activity by suppressing the interaction with Src kinase. In the TX-1918 molecule without the bulky t-butyl groups of TX-1123, the mobility of the phenolic hydroxy group increased, indicating reaction characteristics different from those of TX-1123 by an interaction with Phe405 in Src kinase.
The motility (flexibility) of TX-1926 was altered by the addition of a six-membered ring to the 1,3-dicyclopentenedion site, and the anti-kinase activity decreased. In tyrphostin AG17, the 1,3-cyclopentenedione site was substituted for the cyano group, and the molecular frame was altered. The structural characteristics of tyrphostin AG17 were altered more than those of TX-1123, and not only the phenolic hydroxy group, but also the cyano group interacted with the Src kinase molecule. TX-1927 had a molecular backbone to similar that of tyrphostin AG17, and reactivity with Src kinase was not affected by the methylation of the phenolic hydroxy group. This result did not contradict Src kinase inhibitory activity not being significantly influenced by the methylation of the phenolic group in the TX-1925 molecule.
It appears to be possible to develop drugs with anticancer activity based on the control of protein kinase function, even with small molecules such as members of the TX-1123 family, by using a simulation of molecular interaction with a target protein kinase.
Acknowledgements
This work was supported, in part, by Grant-in-Aid for Scientific Research (15K08113) from the Ministry of Education, Culture, Sports, Science and Technology of the Japanese Government.
- Received April 5, 2016.
- Revision received May 18, 2016.
- Accepted May 24, 2016.
- Copyright© 2016 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Molecular Aspects of C-glycosides: Interactive Analysis of C-linked Compounds With the SGLT2 Molecular Model
- Effect of Isomerization of TX-2036 Derivatives on the Interaction With Tyrosine Kinase Domain of EGF Receptor
- Interactive Analysis of TX-1123 with Cyclo-oxygenase: Design of COX2 Selective TX Analogs