


Abstract
Background: PDZ-binding kinase/T-cell-originated protein kinase (PBK/TOPK) is a serine-threonine kinase and overexpressed in various types of cancer. PBK/TOPK is associated with tumor cell development and progression through suppression of p53 function. In this study, we tested whether PBK acts as a cancer-promoting factor by being overexpressed in esophageal squamous cell carcinoma (ESCC). Materials and Methods: We analyzed PBK/TOPK expression in 15 ESCC cell lines, and 54 primary ESCC tumors that were curatively resected between 1994 and 2007. Results: Overexpression of the PBK/TOPK protein was detected in 93% (14/15) ESCC cell lines and 19% (10/54) primary ESCC tumor samples, and significantly correlated with macroscopic appearance and tumor depth. PBK/TOPK positivity was independently associated with worse outcome in multivariate analysis (p=0.0235, hazard ratio=3.58). Knockdown of PBK/TOPK using specific siRNAs inhibited the cell proliferation, invasion/migration of PBK/TOPK-overexpressing ESCC cell lines. Conclusion: These findings suggest that PBK/TOPK plays a crucial role in tumor malignant potential through its overexpression in ESCC.
Esophageal cancer is the eighth most common cancer in the world (1) and esophageal squamous cell carcinoma (ESCC) accounts for 90% of esophageal carcinomas diagnosed in Asian countries. Although surgical techniques and perioperative management have progressed, ESCC remains one of the most aggressive carcinomas of the gastrointestinal tract. Since finding molecular targets for ESCC treatment might help improve the survival of patients with this lethal disease, studies have attempted to identify biological factors involved in the malignant potential of ESCC. However, few genes have been demonstrated to be associated with biological or pathological features of ESCC, suggesting that novel genes associated with the progression of ESCC need to be identified.
Because identifying molecular targets for ESCC treatment may contribute to the improvement of survival of patients with this lethal disease, several recent studies have clarified that certain molecules, such as cyclo-oxygenase 2 (COX2), B-cell lymphoma (BCL2), tumor protein P53 (TP53), p16, cyclin D1, FAS, epidermal growth factor receptor (EGFR), vascular endothelial growth factor (VEGF), and E-cadherin (2-6), have important roles in tumorigenesis and the development of ESCC. Moreover, various genetic and epigenetic alterations that contribute to the carcinogenesis of ESCC have been elucidated. In clinical settings, however, only a few molecules have been validated as diagnostic, therapeutic, or prognostic biomarkers for ESCC. These findings prompted us to further search for novel cancer-associated molecules.
PDZ binding kinase/T-LAK cell originated protein kinase (PBK/TOPK) encodes a serine/threonine protein kinase and is highly expressed in various types of human cancer such as breast and lung cancer (7-9). Physiologically, PBK/TOPK plays a positive regulatory role in proper chromosomal separation and cytokinesis through phosphorylation of various targets (10, 11). PBK/TOPK gene expression is regulated by the cell-cycle-specific transcription factors E2F and cAMP-responsive element binding/activating transcription factor (CREB/ATF) (12). PBK/TOPK prevents cancer cell death by impairing processes in the DNA damage-induced apoptosis pathway (13-15), such as tumor suppressor p53 and p38-mitogen-activated protein kinase (p38-MAPK) activities. PBK/TOPK promotes cell migration by modulating phosphoinositide 3-kinase/phosphatase and tensin homolog/AKT (PI3K/PTEN/AKT)-dependent signaling (8). However, to date, there has been no report on the clinical and prognostic significance of PBK/TOPK in patients with ESCC and its molecular function contributing to gastric carcinogenesis.
Materials and Methods
Cell lines and primary tissue samples. A total of 15 ESCC cell lines were used, of which 12 belonged to the TE series provided by the Cell Resource Center for Biomedical Research, Institute of Development, Aging and Cancer, Tohoku University and three were KYSE series lines established from surgically resected tumors (16). All ESCC cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (Sigma, St. Louis, MO, USA) supplemented with 100 ml/l fetal bovine serum (Trace Scientific, Melbourne, Australia). All cell lines were cultured in 50 ml/l carbon dioxide at 37°C in a humidified chamber.
Primary tumor samples of ESCC were obtained from 54 consecutive patients with ESCC who had undergone curative esophagostomy at the Division of Digestive Surgery, Department of Surgery, Kyoto Prefectural University of Medicine (Kyoto, Japan) between 1994 and 2007. Each sample was embedded in paraffin after 24 h of formalin fixation. Relevant clinical and survival data were available for all patients. Written consent was always obtained in the formal style and after approval by the local Ethics Committee. None of these patients had undergone endoscopic mucosal resection, palliative resection, preoperative chemotherapy or radiotherapy, and none of them had synchronous or metachronous multiple cancer in other organs. Disease clinical and pathological stage was defined in accordance with the International Union Against Cancer tumor-lymph node-metastases (TNM) classification (17). The median follow-up period for surviving patients was 59.7 months (ranging from 4.5 to 156.9 months).
Quantitative real-time reverse transcription polymerase chain reaction (RT-PCR). Single-stranded complementary DNA generated from total RNA was amplified with primers specific for each gene, as described below. The abundance of messenger RNA (mRNA) was measured with a quantitative real-time fluorescence detection method (ABI StepOnePlus™ Sequence Detection System; Applied Biosystems, Foster City, CA, USA) using TaqMan Gene Expression Assays (Hs00902992_m1 for PBK/TOPK, Applied Biosystems) according to the manufacturer's instructions. The total RNA of normal organs was purchased from Takara Bio Inc., Shiga, JAPAN [Human Total RNA Master Panel II (cat. no. 636643) and Human Liver Total RNA (cat. no. 636531)] and BioChain Institute Inc., Newark, CA, USA (Human Esophagus Total RNA (cat. no. R1234106-50)]. The results of gene expression were calculated as the ratio between PBK/TOPK and expression of the internal reference gene [Hs01060665_g1 for β-actin (ACTB); Applied Biosystems] which provides a normalization factor for the amount of RNA isolated from a specimen. This assay was performed in duplicate for each sample.
Western blotting. Mouse polyclonal antibody against PBK/TPOK (sc-293028) and ACTB antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Cells were lysed and their proteins were extracted by M-PER® Mammalian Protein Extraction Reagent (Thermo Scientific, Waltham, MA, USA).
Immunohistochemistry. Primary tumor samples were fixed with 10% formaldehyde in phosphate-buffered saline (PBS) and routinely embedded in paraffin. The horseradish peroxidase (HRP) staining method was performed. In brief, after deparaffinization, antigen retrieval was performed by heating the samples in 10 mmol citrate buffer (pH 6.0) at 95°C for 1 h. Endogenous peroxidases were quenched by incubating the sections for 20 min in 3% H2O2. After treatment with Block Ace (Dainippon Sumitomo Pharmaceutical, Osaka, Japan) for 30 min at room temperature, sections were incubated at room temperature for 1 h with anti-PBK/TOPK (1:500). PBS was used for all dilutions and washings. The bound primary antibody was detected with EnVision™+ dual link System-HRP (Dako North America, Inc., Carpinteria, CA, USA). HRP labeling was visualized using color development with diaminobenzidine tetrahydrochloride. Slides were counterstained with Mayer's hematoxylin. A formalin-fixed ESCC cell line overexpressing PBK/TOPK (KYSE70 cells), in which >50% of cells showed staining of PBK/TOPK protein, was used as a positive control, whereas KYSE70 cells incubated without the PBK/TOPK antibody were used as a negative control.
For scoring PBK/TOPK expression, the intensity (intensity score: 0=negative, 1=weak, 2=moderate, 3=strong) and percentage of the total cell population (proportion score: 0≤1 <20%, 20% ≤2 <40%, 40% ≤3 ≤100%) that expressed PBK/TOPK was evaluated for each case. Expression of PBK/TOPK was graded as high (intensity plus proportion scores ≥5 of tumor cells showing immunopositivity) or low (intensity plus proportion scores ≤4 of tumor cells showing immunopositivity) using high-power (×200) microscopy (18).
Loss-of-function by small interfering RNA (siRNA) and cell growth analysis. For loss-of-function analysis by knocking down endogenous gene expression, small interfering RNAs (siRNAs) targeting PBK/TOPK (Stealth RNAi™ siRNA #HSS183331; Invitrogen Corporation, Carlsbad, CA, USA), and luciferase (Luc) 5’-CGUACGCGGAAUACUUCGA-3’ (Sigma, Tokyo, Japan) was transfected into TE14 and KYSE70 cells (10 nmol) using Lipofectamine RNAiMAX (Invitrogen Corporation) according to the manufacturer's instructions. The knockdown of the target gene was confirmed by western blotting.
For measurements of cell growth, the number of viable cells at different time points after transfection was assessed by the colorimetric water-soluble tetrazolium salt (WST) assay (Cell counting kit-8; Dojindo Laboratories, Kumamoto, Japan). Cell cycle phase was investigated 72 h after transfection by fluorescence-activated cell sorting (FACS) as described elsewhere (19).
Analysis of apoptotic cells. Cells were treated with staurosporine (200 nmol), which induces intrinsic apoptosis via the activation of caspase-3, for 24 h. Apoptotic cells were determined using an Annexin V-FITC kit (Beckman Coulter, Tokyo, Japan), which contained fluorescein isothiocyanate (FITC)-conjugated annexin V and propidium iodide (PI), as directed by the manufacturer. Apoptotic cells were analyzed by flow cytometry with BD Accuri C6 (BD Biosciences, Tokyo, Japan).
Transwell migration and invasion assays. Transwell migration and invasion assays were carried out in 24-well modified Boyden chambers (transwell-chamber; BD Transduction, Franklin Lakes, NJ, USA). The upper surface of 6.4 mm diameter filters with 8-μm pores was precoated with (invasion assay) or without (migration assay) Matrigel (BD Transduction). The siRNA transfectants (8×105 cells per well) were transferred into the upper chamber. Following 22 h of incubation, the migrated or invasive cells on the lower surface of the filters were fixed and stained with Diff-Quik stain (Sysmex, Kobe, Japan), and stained cell nuclei were counted directly in triplicate. We assessed the invasive potential according to the protocol of Corning® BioCoat™ Matrigel® Invasion Chamber (cat. no. 354480; Corning, New York, NY, USA) by calculating the percentage invasion through the Matrigel-coated filters relative to the migration through the uncoated filters of test cells over that in the control counterparts as described elsewhere (20-22).
Statistical analysis. Clinicopathological categorical variables pertaining to the corresponding patients were analyzed for significance by the chi-square test or Fisher's exact test. Differences of non-categorical variables between subgroups were tested with the non-parametric Mann–Whitney U-test. For the analysis of survival, Kaplan–Meier survival curves were constructed for groups based on univariate predictors, and differences between the groups were tested with the log-rank test. Univariate and multivariate survival analyses were performed using the likelihood ratio test of the stratified Cox proportional hazards model. Differences were assessed with a two-sided test and considered significant at the p<0.05 level.
Results
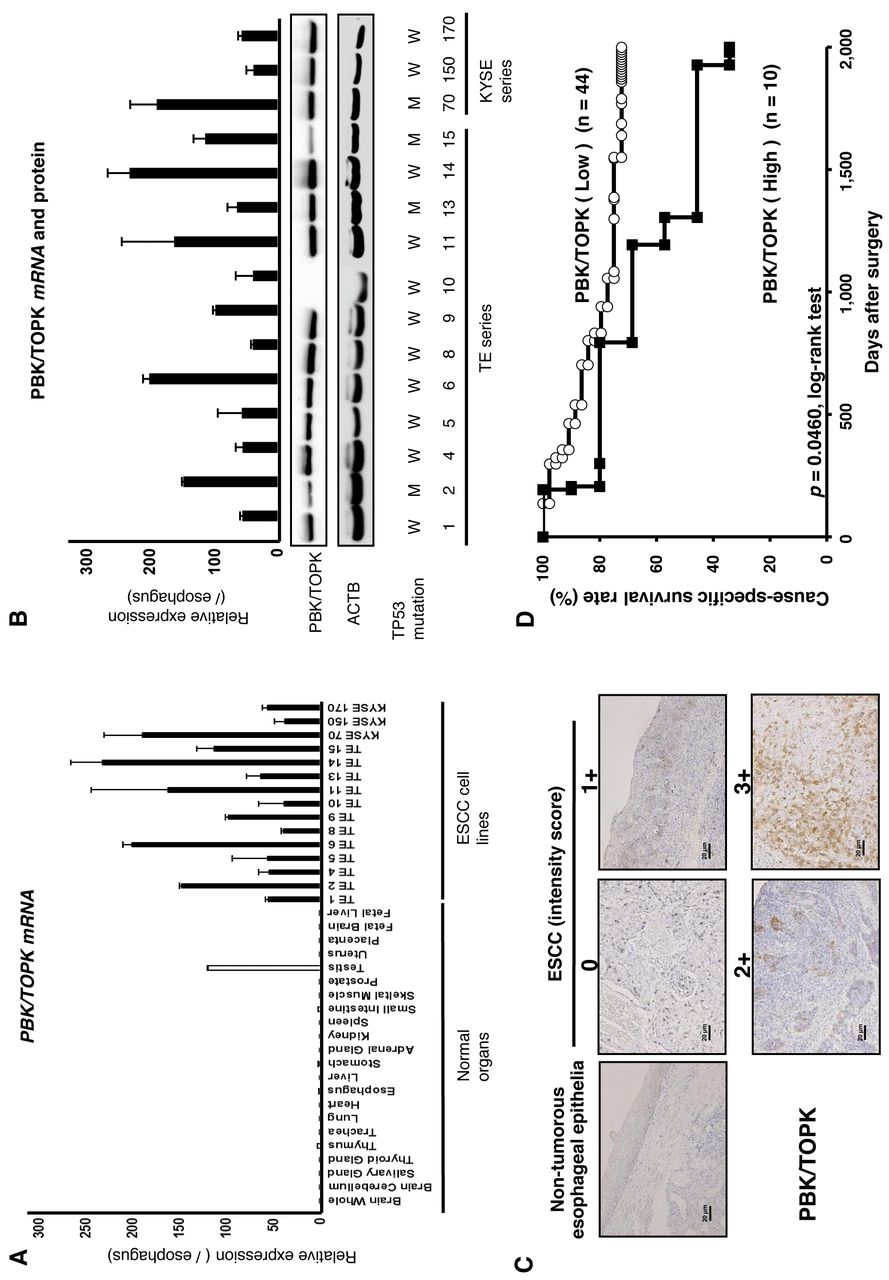
Overexpression of PBK/TOPK in ESCC cell lines. Quantitative RT-PCR analysis was performed to test whether PBK/TPOPK is overexpressed in ESCC cell lines compared to normal organs (Figure 1A). PBK/TPOPK mRNA overexpression was observed in all ESCC cell lines and testis compared to other normal organs, suggesting this gene to be a cancer-testis antigen and a target for activation in ESCC. Expression of PBK/TOPK protein detected by the PBK/TOPK-specific antibody (14/15 cell lines, 93.3%) correlated with that of PBK/TOPK mRNA in ESCC cell lines (Figure 1B). The status of TP53 mutation in the cell lines is reported in the database (http://p53.free.fr/index.html). Although wild-type TP53 may be expected to be a more suitable background than mutant-type TP53 for PBK/TOPK, expression of PBK/TOPK mRNA and protein was not correlated with the mutation status and expression of p53 (Figure 1B).
Distribution of patients of intensity and proportion score.
Immunohistochemical analysis of PBK/TOPK expression in primary tumors of ESCC. Because the PBK/TOPK protein was overexpressed in almost all ESCC cell lines, it was hypothesized that PBK/TOPK was also highly expressed in ESCC tissues and would be associated with carcinogenesis and malignant outcomes. We examined the prognostic and clinicopathological significance of PBK/TOPK expression in primary tumor samples of ESCC based on the immunohistochemical staining pattern of this protein. PBK/TOPK protein was observed in both the cytoplasm and nucleus of cancer cells (Figure 1C). We classified 54 ESCC samples into high- and low-expression groups according to the intensity and proportion of PBK/TOPK staining among tumor cells as described in the Materials and Methods section. In primary cases, PBK/TOPK protein expression was negative in the non-tumorous esophageal epithelia (Figure 1C), and was weakly detected in the lymphoid follicles. Table I shows the distribution of patients based on the intensity and proportion scores for PBK/TOPK immunoreactivity of tumor samples. The group with high expression of PBK/TOPK (10/54 patients, 18.5%) with total scores ≥5 had significantly poorer prognosis than the group with low expression (p=0.0460, log-rank test) (Figure 1D).
Association between PBK/TOPK protein abundance and clinicopathological characteristics in primary cases of ESCC. The relationship between the expression of the PBK/TOPK protein and clinicopathological characteristics is summarized in Table II. Higher protein expression of PBK/TOPK was significantly associated with advanced macroscopic appearance and pT stage in the TNM classification and tended to be associated with advanced pStage in the TNM classification, whereas other characteristics including histological grade were not.
In the Cox proportional hazard regression model (Table III), univariate analysis demonstrated that higher PBK/TOPK protein expression, larger tumor size, venous invasion and lymphatic invasion were significantly correlated with cause-specific survival. When data were stratified for multivariate analysis using both the forward and backward stepwise Cox regression procedures, PBK/TOPK immunoreactivity in tumor cells remained significant (hazard ratio=3.58, 95% confidence interval=1.20-9.65; p=0.0235) for cause-specific survival in the whole patient cohort, suggesting that immunoreactivity for PBK/TOPK may be an independent predictor of cause-specific survival.
Association between clinicopathological characteristics and PDZ-binding kinase/T-cell-originated protein kinase (PBK/TOPK) expression.
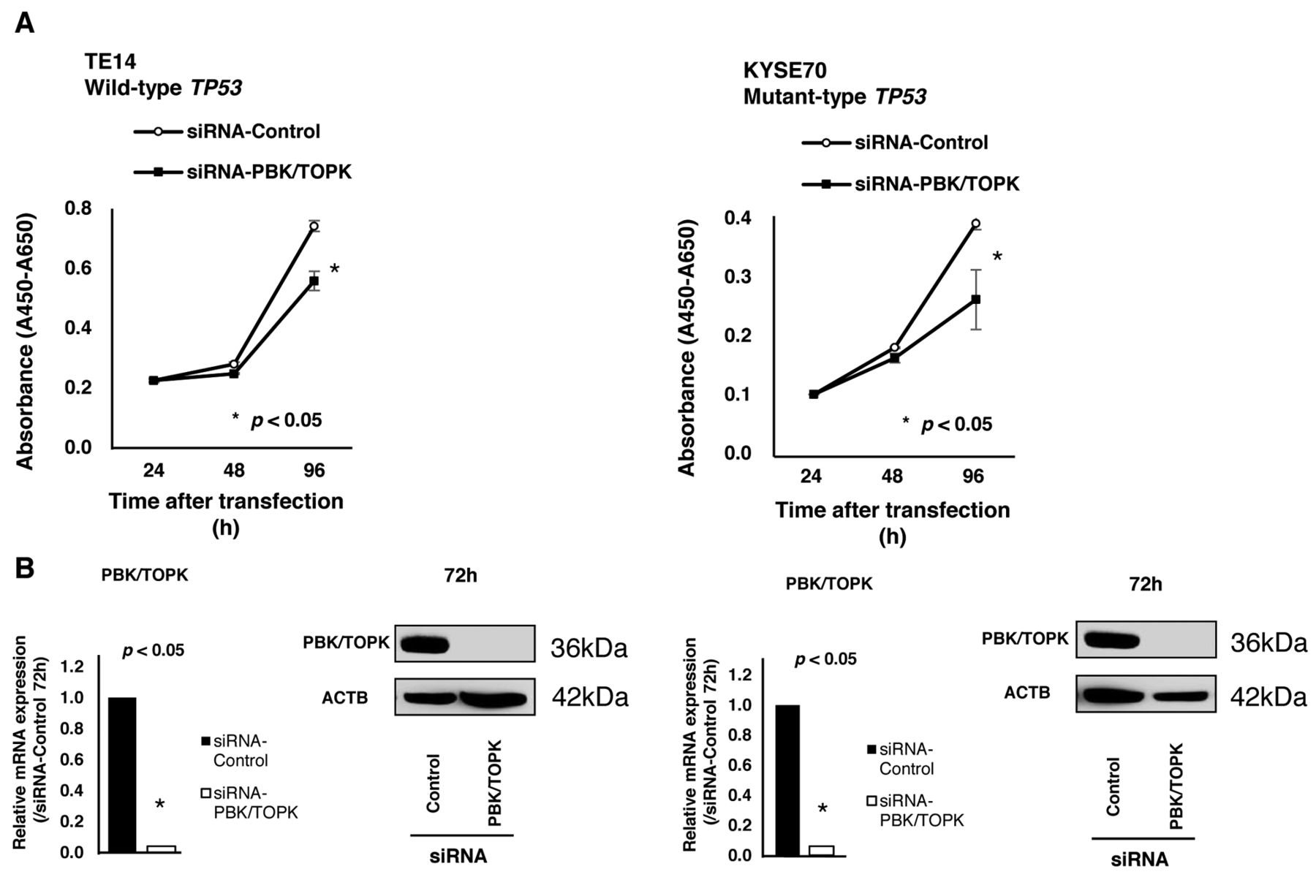
Suppression of cell proliferation by knockdown of PBK/TOPK and the knockdown effect according to TP53 mutation status. To gain an insight into the potential role of PBK/TOPK as an oncogene whose overexpression could be associated with esophageal tumorigenesis, we first performed a cell proliferation assay using several siRNAs specific to PBK/TOPK to investigate whether knockdown of PBK/TOPK would suppress proliferation of ESCC cells that overexpress PBK/TOPK. In wild-type TP53 cell lines, such as TE14 (Figure 2A) and in mutant-type TP53 cell lines, such as KYSE70 (Figure 2B), expression of the PBK/TOPK protein was efficiently knocked-down 72 h after the transient introduction of a PBK/TOPK-specific siRNA compared to the control siRNA (Figure 2A). The proliferation of all these cell lines was significantly lower than that of the controls after the knockdown of endogenous PBK/TOPK expression.
Cell-cycle analysis by down-regulation of PBK/TOPK expression using fluorescence-activated cell sorting (FACS). FACS analysis demonstrated that transfection of wild-type TP53 TE14 cells with siRNA-PBK/TOPK resulted in an accumulation of cells in the G0-G1 phase compared to transfection with control siRNA (Figure 3A). In mutant-type TP53 KYSE70 cells, however, there was an accumulation of cells in the sub-G1 phase compared to transfection with control siRNA (Figure 3B). These findings suggested that PBK/TOPK might be related to cell proliferation according to TP53 status.
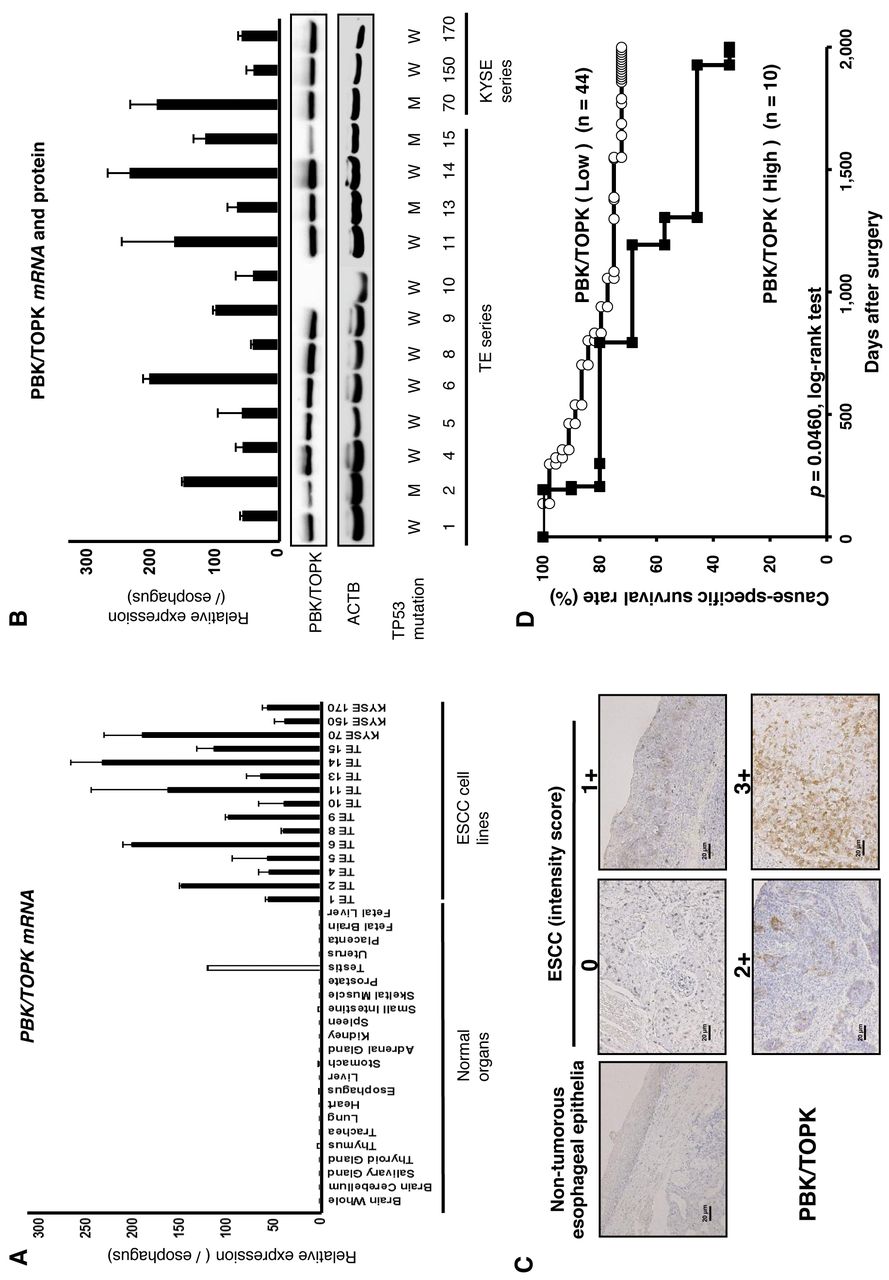
A: Expression of PDZ-binding kinase/T-cell-originated protein kinase (PBK/TOPK) mRNA in 15 esophageal squamous cell carcinoma (ESCC) cell lines compared to that of normal organs. B: Expression of PBK/TOPK mRNA and protein in 15 ESCC cell lines compared to normal esophageal epithelia. The status of TP53 mutation in various cell lines are from the database http://p53.free.fr/index.html. W: Wild-type TP53, M: mutant-type TP53. C: Specific immunostaining of PBK/TOPK in a representative primary tumor sample. Based on this result, the intensity scores for PBK/TOPK staining were determined as follows: 0=negative, 1=weak, 2=moderate, 3=strong. Magnification: ×200, Scale bar: 20 μm. D: Cause-specific survival rates of patients with ESCC as determined by Kaplan-Meier plots according to PBK/TOPK expression determined as high or low.

Effects of PDZ-binding kinase/T-cell-originated protein kinase (PBK/TOPK) knockdown by PBK/TOPK-specific siRNA (siRNA-PBK/TOPK) compared to control siRNA (siRNA-Control) in wild-type TP53 TE14 and mutant-type TP53 KYSE70 cell lines. A: The proliferation of TE14 and KYSE70 cell was significantly lower than with controls after the knockdown of endogenous PBK/TOPK expression. B: The expression of PBK/TOPK protein was efficiently knocked-down 72 h after the transient introduction of a PBK/TOPK-specific siRNA.
Analysis for overall survival.
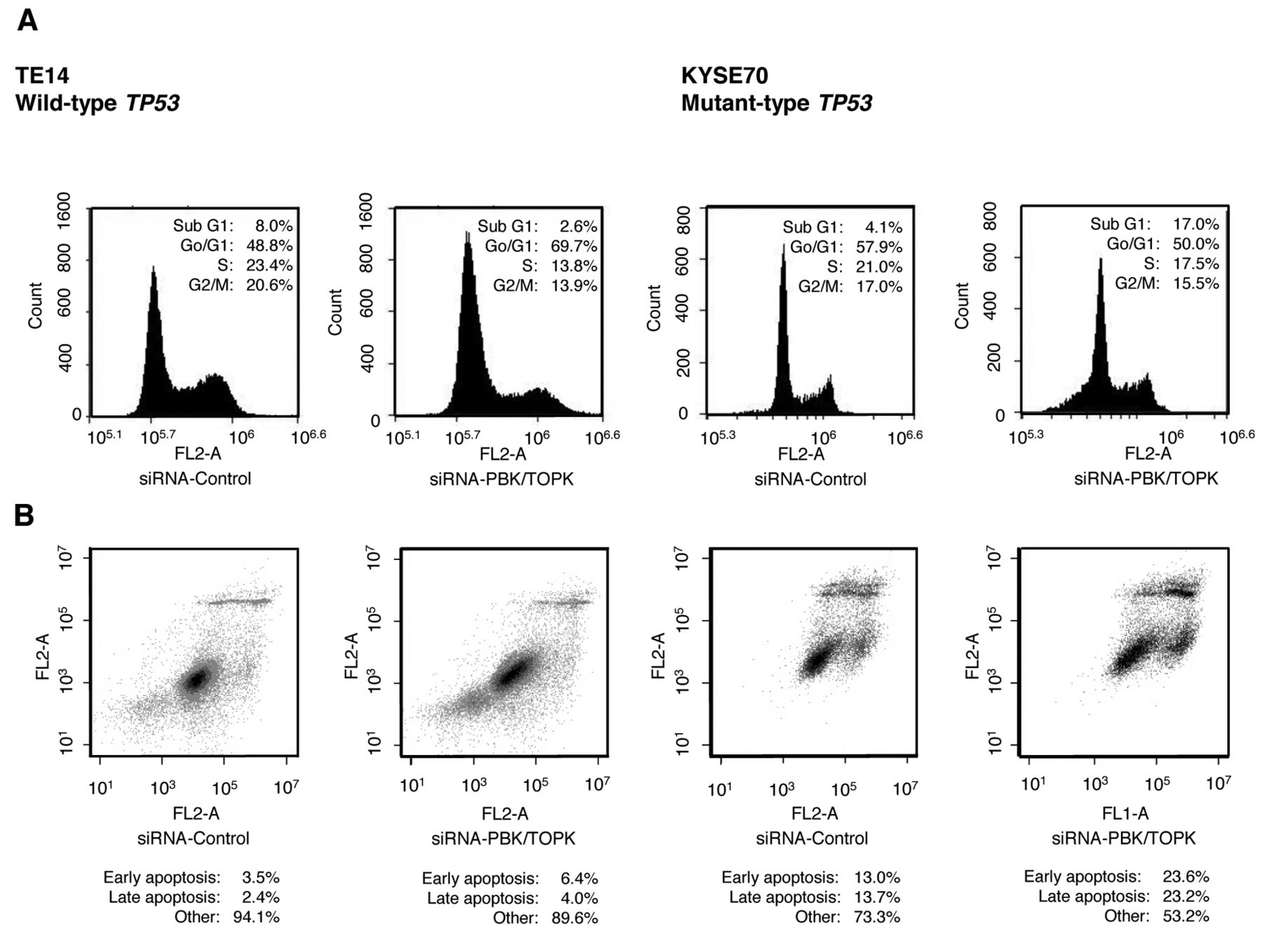
A: Fluorescence activated cell sorting (FACS) analysis demonstrated that transfection of wild-type TP53 TE14 cells with PBK/TOPK-specific siRNA resulted in an accumulation of cells in the G0-G1 phase compared to transfection with control siRNA. In TP53 mutant-type KYSE70 cells, however, there was an accumulation of cells in the sub-G1 phase compared to transfection with control siRNA. B: In apoptotic cell analysis, the down-regulation of PBK/TOPK increased early apoptosis (annexin V-positive/PI-negative) and late apoptosis (annexin V/PI-double positive) 72 h after siRNA-PBK/TOPK transfection compared with transfection with control siRNA in mutant-type TP53 KYSE70 cells, whereas in wild-type TP53 TE14 cells, such an effect was not detected despite the complete knockdown of PBK/TOPK protein.
Analysis of apoptotic cells by knockdown of PBK/TOPK and the knockdown effect according to TP53 mutation status. In order to investigate the accumulation of cells in the sub G1 phase in FACS, apoptotic cell analysis was performed. In mutant-type TP53 KYSE70 cells, the down-regulation of PBK/TOPK increased early apoptosis (annexin V-positive/PI-negative) and late apoptosis (annexin V/PI-double positive) 72 h after siRNA-PBK/TOPK transfection compared to transfection with control siRNA (Figure 3B). In contrast, in wild-type TP53 TE14 cells, accumulation of early or late apoptotic cells was not detected in siRNA-PBK/TOPK-transfected cells compared with control siRNA cells, despite the complete knockdown of PBK/TOPK protein (Figure 3A).
Suppression of cell migration and invasion by down-regulation of PBK/TOPK expression and the knockdown effect according to TP53 mutation status. Next, transwell migration and invasion assays were performed to examine the ability of wild-type TP53 TE14 and mutant-type TP53 KYSE70 cells transfected with siRNA-PBK/TOPK to move through pores under different conditions. Uncoated membrane was used for the migration assays, whereas Matrigel-coated membrane was used for invasion assays. The number of TE14 (Figure 4A) and KYSE70 (Figure 4B) cells that migrated into the lower chamber was significantly lower for siRNA-PBK/TOPK-transfected cells than for siRNA-control-transfected cells under both conditions, suggesting that PBK/TOPK may increase the ability of ESCC cells to migrate and invade in a TP53 mutation-independent manner.

Regarding the cell migration and invasion, transfection with PBK/TOPK-specific siRNA suggests that PBK/TOPK may increase the ability of esophageal squamous cell carcinoma cells to migrate and invade in a TP53 mutation-independent manner. A: TE14 wild-type TP53; B: KYSE70 mutant-type TP53.
Discussion
The PBK/TOPK gene is up-regulated in various types of cancer and tumors such as bladder cancer, brain tumor, breast cancer, liver cancer, lung cancer and sarcoma (7-9, 23-26). PBK/TOPK was initially identified as a mitotic protein kinase (27), assisting the recruitment of cylcin-dependent kinase 1 (CDK1)/cyclin B to mitotic spindles, where PBK/TOPK plays a role in the formation of the spindle midzone and cytokinesis (10). Previous studies have identified various oncogenic functions of PBK/TOPK related to multiple signaling pathways such as the MAPK pathway (14), PI3K/AKT pathway (8, 28, 29) and p53 pathway (13, 15). Through the MAPK pathway, PBK/TOPK functions as mediator of growth-factor activation of p38, with a role in motility and as part of the DNA damage sensing machinery, and is necessary for the phosphorylation of histone H2AX (14). Regarding the PI3K/AKT pathway, PBK/TOPK decreases the PTEN level and regulates PI3K/AKT-stimulated migration (8). Most importantly, by impairing the DNA damage-induced apoptotic pathway, that is the p53 pathway, PBK/TOPK prevents cancer cell death (13). PBK/TOPK physically interacts with p53, specifically through its DNA-binding domain and promotes tumor cell survival and resistance to chemotherapy-induced apoptosis through suppression of p21 (15). Thus, PBK/TOPK has potential oncogenic functions, and these findings prompted us to clarify the clinicopathological and independent prognostic significance of PBK/TOPK overexpression/activation through the p53 pathways in primary ESCC.
Here, we hypothesized that overexpression/activation of PBK/TOPK may promote tumor cell proliferation and lead to poor survival of patients with ESCC. To verify this hypothesis, we firstly examined the expression status of PBK/TOPK in cell lines and primary tumors of ESCC, and its clinicopathological and prognostic significance were evaluated. As a result, we demonstrated that PBK/TOPK was overexpressed in 18.5%, (10/54) of primary ESCCs as well as in 93.3% (14/15) of ESCC cell lines, and this overexpression was an indicator of poor prognosis independently of other prognostic factors. These results suggest that immunoreactivity to PBK/TOPK may be useful as an independent prognosticator in patients with ESCC.
In our in vitro analyses, knockdown of PBK/TOPK overexpression in wild-type TP53 cells induced cell cycle G0-G1 arrest, but its knockdown in mutant-type TP53 cells induced cell cycle sub-G1 accumulation (Figure 3). Moreover apoptotic cell analysis demonstrated that the down-regulation of PBK/TOPK increased early apoptosis (annexin V-positive/PI-negative) and late apoptosis (annexin V/PI-double positive) 72 h after siRNA-PBK/TOPK transfection compared with transfection with control siRNA in mutant-type TP53 TE14 cells (Figure 3B). This suggests that in tumors with wild-type TP53, p53 may be an important background for PBK/TOPK, as reported previously (15). In p53-mutant patients, however, it suggests that PBK/TOPK may contribute to tumorigenesis by suppressing apoptosis in a pathway different from the p53 pathway.
Next, we investigated further the molecular mechanism affecting the malignant potential in tumor cells with both overexpression of PBK/TOPK and mutant-type TP53. As a result, knockdown of PBK/TOPK suppressed invasion and migration in mutant-type as well as wild-type TP53 ESCC cells (Figure 4). Thus, the in vitro findings strongly supported our in vivo results of immunohistochemical analysis and strongly suggest that PBK/TOPK plays a pivotal role in the malignant potential of ESCC.
Recently, PBK/TOPK-specific inhibitors such as OTS514, OTS964 (OncoTherapy Science Inc., Japan) (30) and HI-TOPK-032 (24, 31) have been developed. OTS964 was shown to be extremely effective in treating lung cancer in a xenograft mouse model, without showing any adverse reactions (30). Regarding p53 status, Kim et al. reported that the administration of PBK/TOPK-specific inhibitor HI-TOPK-032 suppressed tumor growth in a wild-type TP53 HCT116 colon cancer xenograft model (24, 31). Ikeda et al. more recently reported that oral administration of OTS514 significantly increased overall survival in TP53 mutant-type ES-2 abdominal dissemination xenograft model, compared to vehicle control in ovarian cancer (32). These data indicate that the molecular targeting of PBK/TOPK may work for tumors of both wild-type and mutant-type TP53 in various cancer types. Therefore, PBK/TOPK-specific inhibitors may be key molecules for the treatment of patients with PBK/TOPK-overexpressing ESCC. These inhibitors are currently being evaluated in vitro and in vivo at our institute.
In conclusion, as far as we are aware, this is the first report to show that PBK/TOPK has a pivotal oncogenic role through molecular mechanisms associated with the p53 and is a potential therapeutic target in ESCC. We clearly demonstrated the frequent overexpression of PBK/TOPK protein and its prognostic value in patients with ESCC. Although studies of larger cohorts are needed to validate these findings before moving to clinical settings, our results provide evidence that PBK/TOPK could be a crucial molecular marker for determining the malignant properties of ESCC cells and that it could be a target for molecular therapy in patients with ESCC.
Footnotes
↵* These Authors contributed equally to this study.
This article is freely accessible online.
- Received October 2, 2016.
- Revision received October 30, 2016.
- Accepted October 31, 2016.
- Copyright© 2016 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}