


Abstract
Background/Aim. The pancreatic cancer cell line KLM1 can gain chemoresistance following gemcitabine (GEM) treatment. Metformin was found to be a useful sensitising agent towards GEM treatment following gain of chemoresistance. Materials and Methods: The proliferation of GEM-sensitive and -resistant cells was investigated over a range of metformin concentrations from 0.005 to 5 mM. The intra- and extra-cellular energetic profiles of these two cell types under metformin exposure were investigated through adenosine triphosphate (ATP) and L-lactate assays. Results: There was an unexpected decrease in intracellular L-lactate following gain of chemoresistance, despite observable medium acidification. At the biochemical level, a mar ked effect on phosphorylated proteins upstream of Akt, along the mTOR pathway, was observed at 6 h. These changes followed a time-dependent pattern linked closely to the changes in the energetic profile. Conclusion: Together, these results indicate that metformin indirectly blocks protein phosphorylation, including that of heat shock protein 27 (HSP27).
Metformin (N,N-Dimethylimidodicarbonimidic diamide), a biguanide, is a widely used drug by type II diabetics and has been linked to a lower cancer incidence as shown in diabetics treated with metformin compared to those on other diabetic drugs (1-5).
Metformin was reported to indirectly activate adenosine monohosphate (AMP)-activated protein kinase (AMPK) activity by disrupting complex I of the mitochondrial respiratory chain and subsequently adenosine triphosphate (ATP) synthesis (6-8), which leads to phosphorylation by liver kinase B1 (LKB1) (9). This brings about a reduction in the mammalian target of rapamycin (mTOR) (10-14) and an associated downstream signalling suppression, such as eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), ribosomal protein S6 kinase 1 (S6K1) and human epidermal growth factor receptor 2 (HER-2) (9, 11, 13). The suppression of mTOR also modulates other targets (13, 15-23) including the activation of tuberous sclerosis complex 2 (TSC2, tuberin), which further suppresses mTOR activity (24). Dysregulation of mTOR is common in many cancers and mTOR is known to be central in the phosphatidy linositol-3-kinase/ protein kinase B/mTOR (PI3K/Akt/mTOR) signalling pathway (25, 26).
Metformin has been found to also enhance apoptosis induction via the tumor suppressor p53, which is activated by AMPK (27, 28). Additionally, it suppresses cyclin D1 and E2F1 expression leading to decreased cancer prolife ration (14, 20), as well as a variety of mitosis-related gene families of which tubulins, histones and aurora kinases are of particular interest (18).
This study was designed to understand the effect of metformin on the pancreatic cancer cell lines KLM1 and KLM1-R particularly focusing on the effect on heat shock protein 27 (HSP27) in these two cell lines. Although many cancer types have been tested with metformin, it has only been recently demonstrated to suppress pancreatic cancer proliferation in patients and cell lines (ASPC-1, BxPc-3, PANC-1 and SW1990) (29, 30). HSP27 is known to associate with Akt (31, 32), which is activated via PI3K and stalls apoptosis (33, 34) by phosphorylating pro-apoptotic factors, such as B-cell lymphoma-2 (BCL-2)-associated death protein (BAD), leading to a sequestration and inhibition by 14-3-3 proteins (35). HSP27 has been shown to have the ability to reduce apoptosis by blocking the truncated BH3 interacting domain death agonist (tBID), as well as reduce the SMAC and cytochrome C release from mitochondria (36).

Metformin brings about a marked decrease in the proliferation of the Gemcitabine-resistant pancreatic cell line KLM1-R, comparable to its Gemcitabine-sensitive parent cell line KLM1 within the first 48h of treatment.
Materials and Methods
Pancreatic cancer cell lines and culture conditions. The cell lines used were KLM1 (provided by the Department of Surgery and Science at Kyushyu University Graduate School of Medical Science), KLM1-R (gemcitabine resistant KLM1) (37) (provided by the Department of Surgery and Science at Kyushyu University Graduate School of Medical Science), MIA PaCa-2 (provided by the Institute of Development, Aging and Cancer at Tohoku University) and PK-59 (provided by the Institute of Development, Aging and Cancer at Tohoku University). They were cultured in Dulbecco's modified Eagle's medium (DMEM) (high glucose; Wako, Osaka, Japan) with 2mM L-glutamine and 10% FBS at 37°C, in a 5% CO2 atmosphere, at high relative humidity.
Proliferation assays. KLM1 and KLM1-R cells were cultured in 96-well plates and treated with metformin at varying concentrations (from 0.005-5 mM) in conjunction with 500 ng/ml gemcitabine (GEM; Eli Lilly, Indianapolis, IN, USA). Cell proliferation assays using 3-(4,5-dimethyl-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS; CellTiter 96® Aqueous One Solution Cell Proliferation Assay; Promega, Madison, WI, USA) were carried out at 24, 48 and 72 h to determine the ability of these cell lines to multiply after exposure to metformin treatment. Absorbance readings at 490 nm were taken after 1 h and 2 h incubation with MTS reagent.
ATP and L-lactate assays. KLM1 and KLM1-R cells were cultured in DMEM with and without 10% FBS in 96-well plates and treated with 5 mM metformin for a period of 72 h. Intra- and extra-cellular ATP and L-lactate concentrations were measured at 2-h intervals for the first 8h and then at 24, 48 and 72 h following the start of the experiment using ATP and L-lactate Colorimetric/Fluorometric Assay Kits (BioVision Inc., Milpitas, CA, USA). Absorbance readings at 570 nm were taken after 1h incubation with the appropriate probe.

Metformin reduced cellular proliferation of KLM1 in a dose-dependent manner over the tested range of 0.005-5 mM.
Western blot analysis. KLM1-R cells were cultured in 6-well plates and treated with 5mM metformin for 6h. Cells were lysed using a lysis buffer containing 1 % NP-40, 1 mM sodium vanadate, 1 mM phenylmethanesulfonyl fluoride (PMSF), 10 mM sodium fluoride (NaF), 10 mM ethylenediaminetetraacetic acid (EDTA), 50 mM Tris, 165 mM NaCl, 10 μg/ml leupeptin and 10 μg/ml aprotinin and incubating the cell suspensions for 1h at 4°C with vigorous shaking. The samples were then spun down at 15,000 rpm for 30 min and the supernatants were stored at −80°C until use. After electrophoresis using 12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), proteins were electroblotted onto polyvinylidene fluoride (PVDF) membranes (Immobilon-P; Millipore, Bedford, MA, USA) and blocked for 1 h at room temperature using TBS containing 5% skimmed milk. The primary antibodies used were a mouse monoclonal anti-human HSP27 antibody (sc-13132; 1:1000 dilution; Santa Cruz Biotechnology, Dallas, TX, USA), rabbit polyclonal anti-human pHSP27-Ser15 antibody (#2404; 1:1000 dilution; Cell Signalling Technology, Boston, MA, USA), rabbit polyclonal anti-human pHSP27-Ser78 antibody (#2405; 1:1000 dilution; Cell Signalling Technology, Boston, MA, USA), rabbit polyclonal anti-human pHSP27-Ser82 antibody (#2401; 1:1000 dilution; Cell Signalling Technology, Boston, MA, USA), rabbit polyclonal anti-human Akt antibody (#9272; 1:1000 dilution; Cell Signalling Technology, Boston, MA, USA), rabbit polyclonal anti-human pAkt-Ser473 antibody (#4058; 1:1000 dilution; Cell Signalling Technology, Boston, MA, USA), rabbit polyclonal anti-human AMPK-α1 antibody (#07-350; 1:1000 dilution; Merck Millipore, Hertfordshire, WD, UK), goat polyclonal anti-human pERK-Tyr204 antibody (sc-7976; 1:1000 dilution; Santa Cruz Biotechnology, Dallas, TX, USA), rabbit monoclonal anti-human Src antibody (#2109; 1:1000 dilution; Cell Signalling Technology, Boston, MA, USA), rabbit monoclonal anti-human pSrc-Tyr527 antibody (#2105; 1:1000 dilution; Cell Signalling Technology, Boston, MA, USA), rabbit polyclonal anti-human HSP70 antibody (SPA-812; 1:1000 dilution; Stressgen, San Diego, CA, USA), mouse monoclonal anti-human HSP90 antibody (sc-13119; 1:1000 dilution; Santa Cruz Biotechnology, Dallas, TX, USA), goat polyclonal anti-human GAPDH antibody (sc-20357; 1:1000 dilution; Santa Cruz Biotechnology, Dallas, TX, USA) and a goat polyclonal anti-human Actin antibody (sc-1616; dilution range 1:200; Santa Cruz Biotechnology, Dallas, TX, USA). For each primary antibody, membranes were incubated overnight at 4°C, with shaking. Membranes were then washed three times with TBS containing 0.05% Tween-20 and, finally, with TBS before being incubated with horseradish peroxidase-conjugated secondary antibody (dilution range 1:10,000; Jackson ImmunoResearch Lab., West Grove, PA, USA) for 2h at room temperature, with shaking. Development was carried out using chemifluorescence reagent (ImmunoStar LD; Wako, Osaka, Japan). Immunoreactive protein bands were detected using the Image Reader LAS-1000 Pro (Fujifilm Corporation, Tokyo, Japan).

The effect of metformin treatment is prolonged much beyond the administration period. Metformin was added at a dose of 5 mM for a period of 6 h (spike) or throughout the 72 h experiment duration (constant).
Results
Metformin brings about a marked decrease in proliferation of the pancreatic cell lines KLM1 and KLM1-R. To determine if metformin has an effect on pancreatic cancer, the cell lines KLM1 and KLM1-R, which are GEM-sensitive and resistant, respectively, were chosen since GEM is the main chemotherapeutic agent for this type of cancer. The initial test involved the combination of 500 ng/ml GEM and 5 mM metformin, which is the median of the therapeutic range of action. It is evident that both cell lines are sensitive to this combination to a similar extent. It seems to indicate that metformin sup presses biochemical pathways that nullify the GEM resistance mechanism in KLM1-R cells bringing their proliferation down to the same level as GEM-sensitive KLM1 cells (Figure 1).
The effect observed on cancer proliferation is dose-dependent within the tested range of 0.005-5 mM metformin. In order to understand the effectiveness of metformin over a range of concentrations, doses from 0.005 to 5 mM metformin (in combination with 500 ng/ml GEM) were tested following an incubation of 72 h. The reduction in cell proliferation was found to be directly proportional to the metformin concentration used (Figure 2).

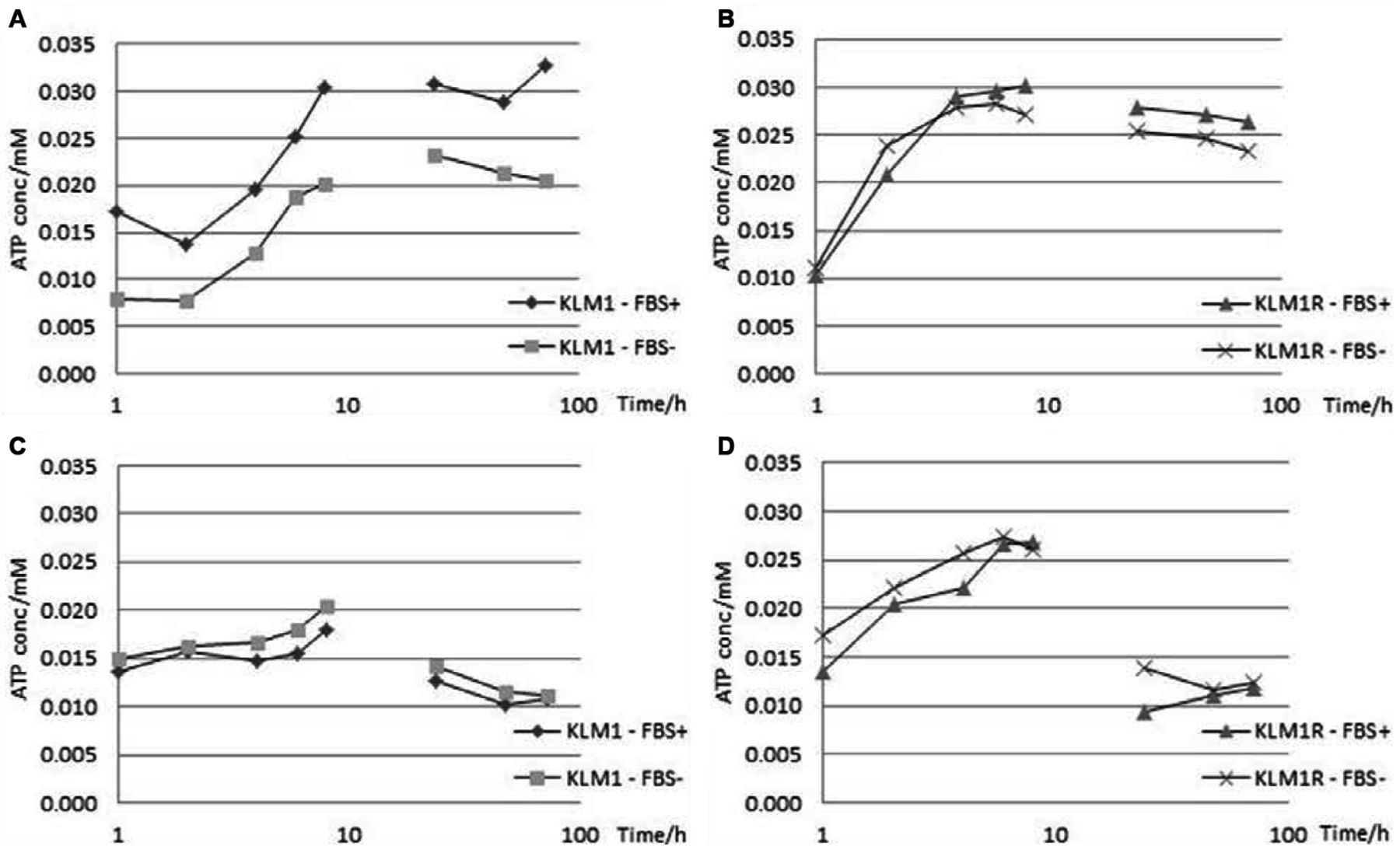
The intracellular (4A, 4B) and extracellular (4C, 4D) L-lactate profiles for KLM1 and KLM1-R under the effect of metformin in medium supplemented with or lacking FBS over a period of 72 h sampled every 2 h for the first 8h and then every 24 h. L-lactate levels appear to undergo an increase intra-cellularly but a decrease extra-cellularly, with the intracellular L-lactate concentration presenting a link to FBS supplementation, most observable in KLM1.

The intracellular (5A, 5B) and extracellular (5C, 5D) ATP profiles for KLM1 and KLM1-R under the effect of metformin in medium supplemented with or lacking FBS over a period of 72 h sampled every 2 h for the first 8 h and then every 24 h. ATP levels appear to undergo a marked initial increase followed by a slow decrease and do not seem to be linked to FBS supplementation.
The effect of metformin treatment is prolonged way beyond the administration period. To study the half-life and effect of different metformin administration strategies, metformin was added at a dose of 5 mM for a period of 6hr (spike) or throughout the entire 72 h duration. The results showed no significant difference between the two set-ups indicating that the initial dose taken-up by cells is enough to reduce prolife ration even if the excess is removed (Figure 3). This suggests that metformin either binds tightly to its target and leads to a gradual decrease in cell proliferation or it is not metabolised and produces an effect through repeated interventions, or else, through some kind of positive feedback loop.
The energetics of KLM1 present a marked shift after becoming chemoresistant as observed through L-lactate and ATP levels. In all proliferation assays, including metformin, it was observed that the metformin-treated samples presented a decrease in pH from 7.4 to 6.1 compared to their equivalent metformin-free controls. For this reason, an assay for L-lactate (both intra- and extra-cellular) was carried-out. A number of interesting observations emerged both in terms of cellular dynamics and effects of experimental conditions.
A peak in intracellular L-lactate level was reached at 24 h, which was dependent on the FBS setting (i.e. presence or absence). However, KLM1-R cells appeared to be much less dependent on FBS and their peak L-lactate concentration was much lower (by almost 50%). Another noteworthy observation is the dip at 6 h, which was observable in both KLM1 and KLM1-R for both FBS settings. The magnitude of the change, however, was positively correlated with the FBS setting, being more pronounced in the presence of FBS (Figure 4A and 4B).
The extracellular L-lactate levels for both KLM1 and KLM1-R cells, however, were almost identical. The only observable difference was the peak obtained at 6 h in KLM, consistent with the dip observed intra-cellularly. This was absent in KLM1-R, possibly indicating that, instead of being secreted, it was being redirected to some biochemical pathway (Figure 4C and 4D).
At the end of the 72 h experimental period, the intracellular L-lactate concentration was higher than that at the start, while the extracellular L-lactate concentration was lower than the initial concentration. In all 4 conditions, the final concentrations of L-lactate show a tendency towards further decrease (Figure 4 A-D).
On the other hand, the peak in intracellular ATP level was reached somewhere between 8 and 24 h, which was dependent on the FBS setting in KLM1. However, KLM1-R cells presented almost overlapping profiles with little dependence on FBS. The other noteworthy observation is the dip at 2h, which was observable only in KLM, in the presence of FBS (Figure 5A and 5B).
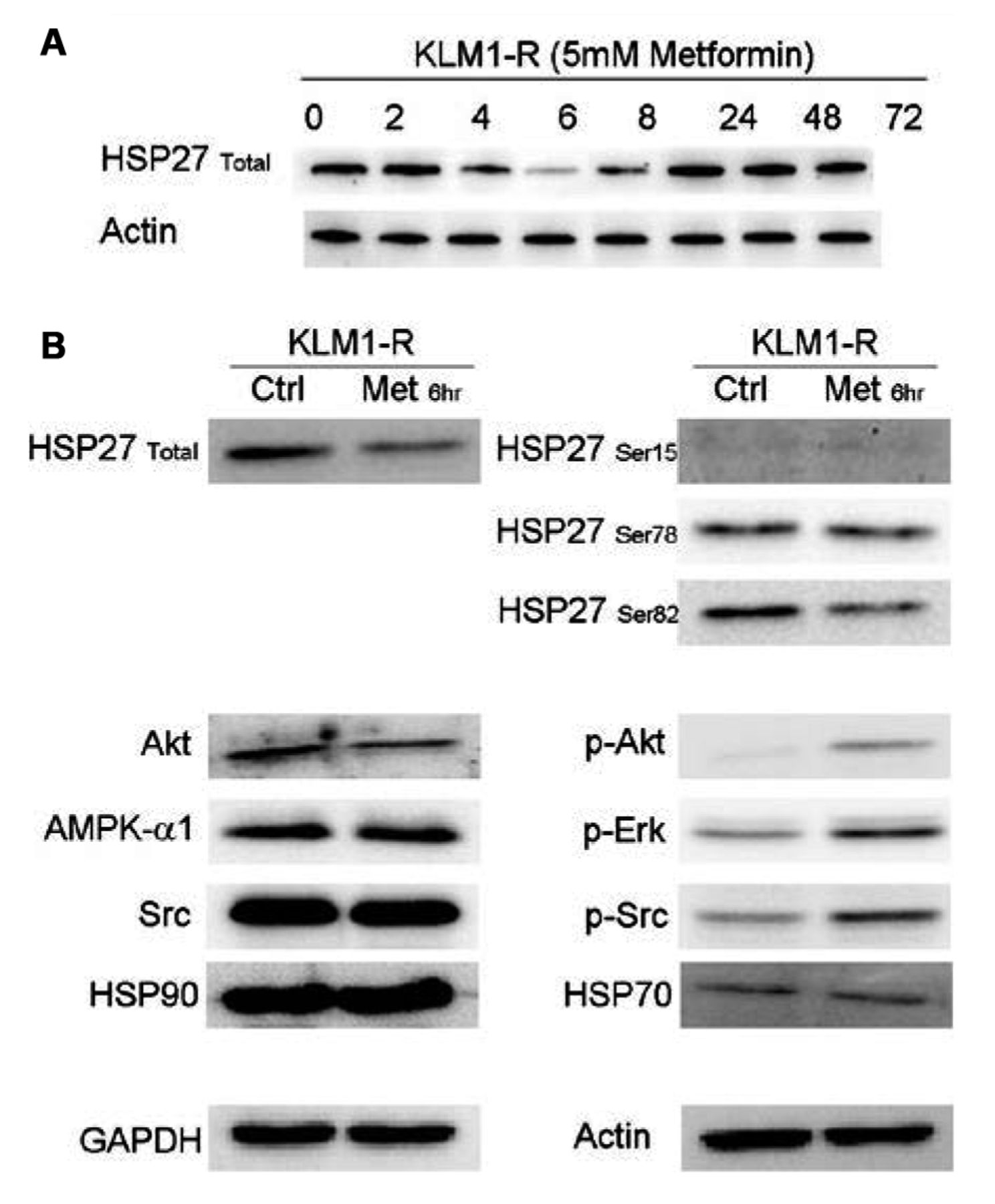
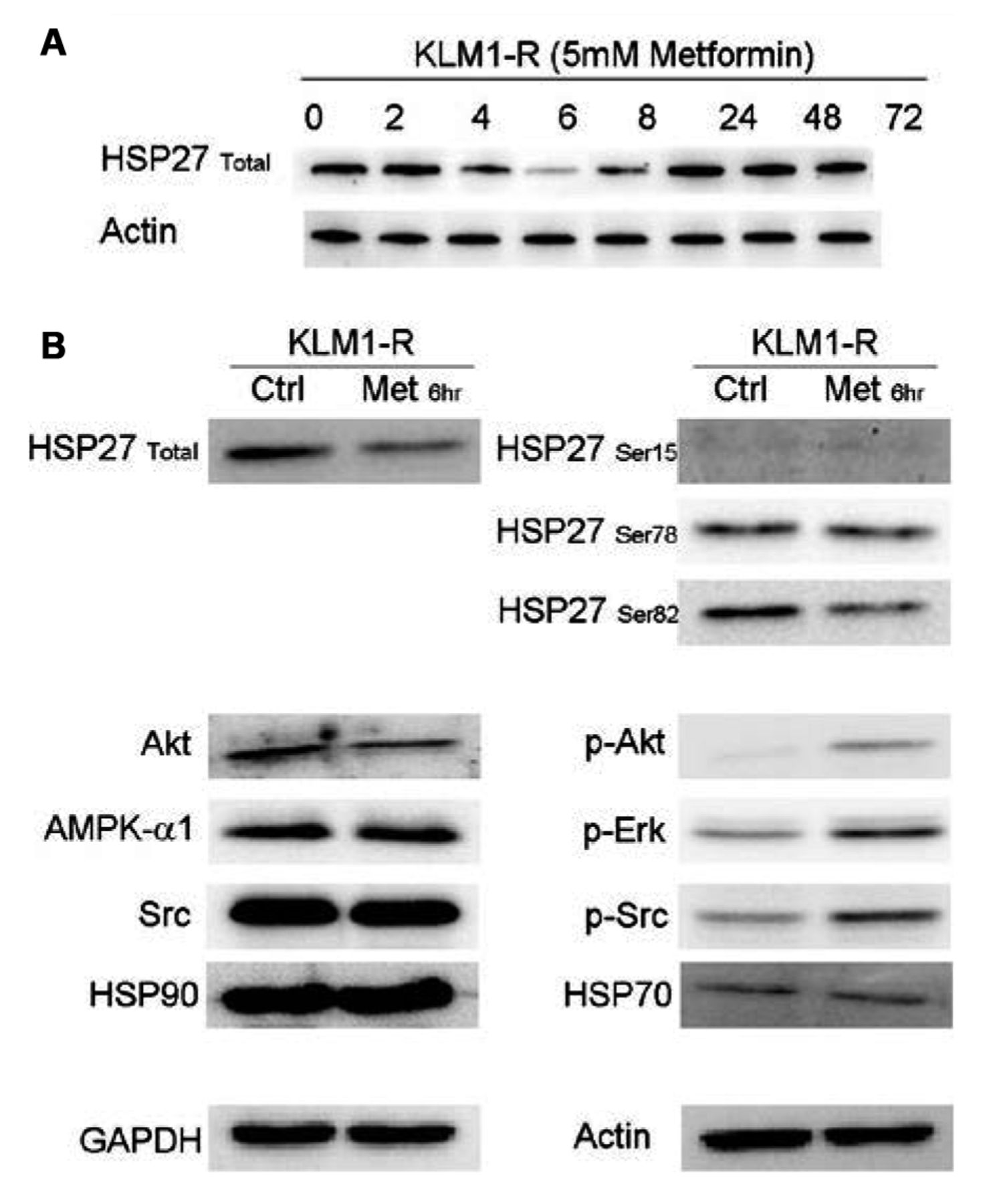
Western blotting experiments. A: HSP27 expression over the first 24 h following metformin administration. B: Expression levels of proteins upstream of and including Akt. The phosphorylation of proteins upstream of Akt is affected by metformin treatment.
The extracellular ATP levels for both KLM1 and KLM1-R cells reached a peak at 8 h. An observable difference was the peak obtained at 2 h in KLM1 in the presence of FBS, consistent with the dip observed intra-cellularly. By 24 h, the ATP level in both KLM1 and KLM1-R cells was lower than that at the start of the experiment (Figure 5C and 5D).
Similar to the L-lactate experiment, at the end of the 72 h experimental period, the intracellular ATP concentration was higher than that at the start, while the extracellular ATP concentration was lower than the initial concentration. In all 4 conditions, the final concentrations of ATP show a tendency towards further decrease (Figure 5 A-D).
Furthermore, the L-lactate and ATP profiles in KLM1 and KLM1-R were subsequently compared to those of GEM-sensitive MIA PaCa-2 and GEM-resistant PK-59 (data not shown). MIA PaCa-2 and PK-59 presented different profiles to KLM1 and KLM1-R, with PK-59 presenting only small fluctuations throughout the experiment. Since the mode of GEM resistance in KLM1-R and PK-59 is through different mechanisms, these results may indicate that the activity of metformin is less effective in PK-59 as this was reflected in the minor alterations observed in the energy profile.
The expression of various phosphorylated proteins upstream of Akt is altered by metformin. Western blotting was carried out for HSP27 over the entire duration of the metformin administration (72 h), sampling at the same time-points as the energetics experiment (Figure 6A). The fluctuation in HSP27 expression appeared to be directly correlated to the L-lactate profile. As the 6 h time-point appeared to be critical in the L-lactate profile, as well as in HSP27 expression, further western blotting analysis was carried out for Src, Erk, HSP27 and HSP90 proteins upstream of Akt (Figure 6B). KLM1-R cells treated with 5 mM metformin for 6 h were used for this experiment. At the 6-h time-point the total HSP27 and phosphrylated HSP27-Ser82 expression was reduced as was that for Akt. In contrast, pAkt, pERK and pSrc levels increased. It appears that, in general, the phosphorylation status of proteins in the mTOR pathway was affected and that this translated into increased sensitisation to GEM and decreased cellular proliferation.
Discussion
Based on previous reports of mTOR pathway involvement in metformin action (10-14) and the over-expression of HSP27 in KLM1 upon gaining resistance to GEM (38), the cell lines KLM1 (GEM-sensitive) and KLM1-R (GEM-resistant) were chosen to study the energetic and biochemical changes observed upon administration of metformin, particularly focusing on the effect on the role played by HSP27 in this type of treatment.
The data gathered fit-in with recently reported mechanistic explanations for the effects of metformin on various cancer types. Based on current understanding, the action of metformin can be described as being two-fold. The first is the inhibition of the mitochondrial electron transport chain complex 1 (ETC1), while the second is activation of AMPK. Via ETC1, metformin acts on AMP/ATP ratios through a decrease in oxidation of reduced nicotinamide adenine dinucleotide (NADH) to nicotinamide adenine dinucleotide+ (NAD+), required to maintain the function of the tricarboxylic acid cycle (TCA) cycle, and various other aspects of mitochondrial activity linked to oxidative stress (39-42). The AMPK pathway on the other hand can be activated in either an LKB1-dependent or independent manner and reduce protein synthesis via activation of tuberous sclerosis complex 1/2 (TSC1/2), which inhibits mTOR (43-45). Therefore, cells treated with metformin undergo a metabolic shift restricting glucose oxidation by mitochondria and opting for glycolysis with increases in purine biosynthesis and TCA cycle intermediates.

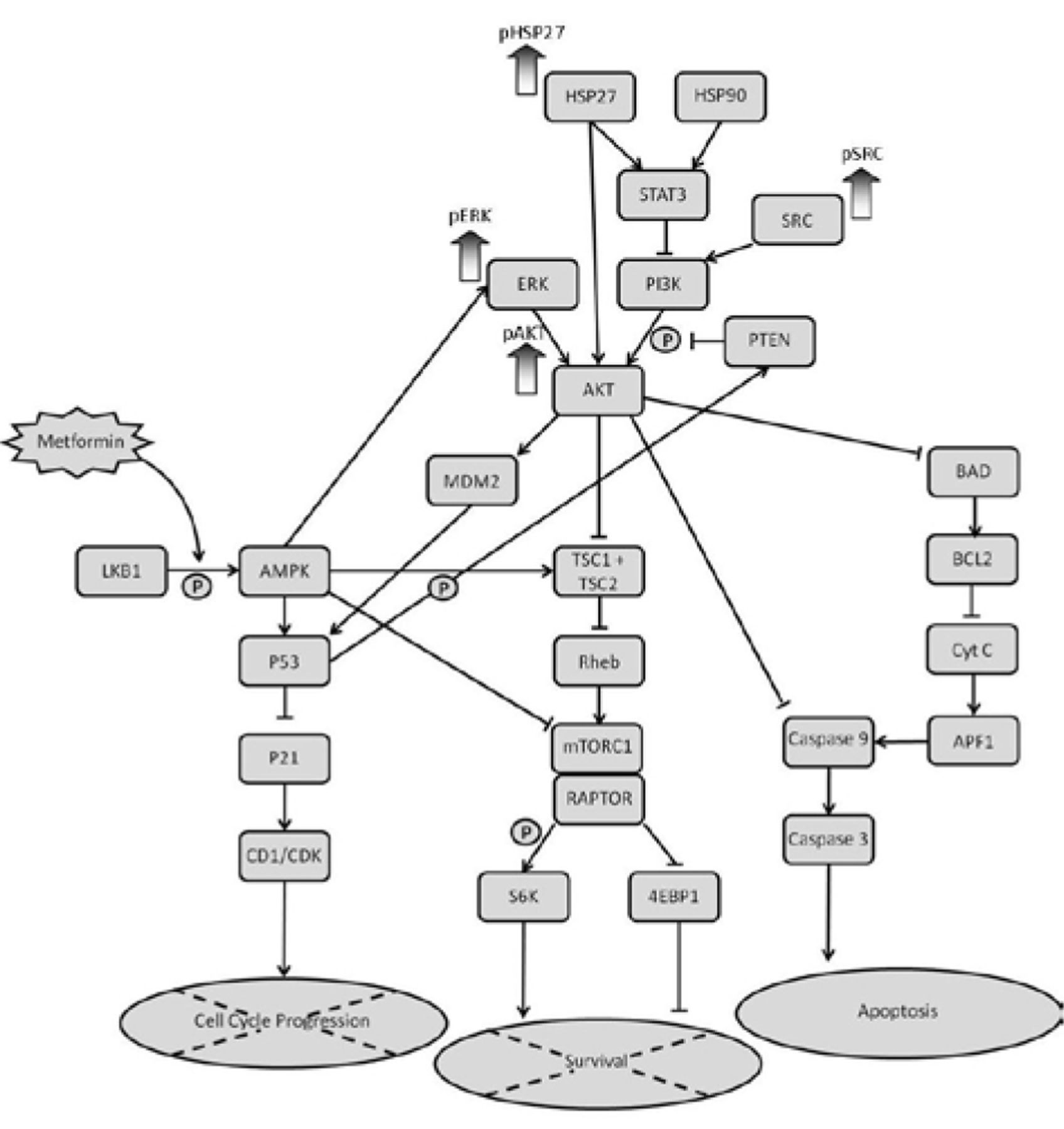
Schematic representation of the mTOR pathway and the proteins that are up-regulated in KLM1-R following treatment with metformin.
The results presented herein confirm that the anti-proliferative and pro-apoptotic effects of metformin do not alter total AMPK levels (although phosphorylation at Thr-172 was no probed for) (20, 43, 46-48). The transient changes observed in biochemistry were also previously reported as an adaptation to the early inhibitory effects of metformin on mitochondrial energetics with a redirection of carbon flux towards L-lactate and other biochemical intermediates (48).
The increase in pAkt level has been previously described as also being part of the above-mentioned adaptive mechanism. It fulfils its role as a key regulator of glucose metabolism by increasing the translocation of glucose trans porters, GLUT1 and GLUT4, to the plasma membrane to increase glucose uptake and aerobic glycolysis (49-51) together with other processes that affect apoptosis (48, 52-55).
It has also been shown that cells with high basal pAkt, glucose consumption or glycolysis are resistant to the early inhibitory effects of metformin and can, thus, avoid apoptosis (48). This does not seem to be an issue with KLM1-R cells, which, as shown by Western blotting, exhibit a very low pAkt expression level.
Recent research has shown that KLM1-R cells over-express SOX2, a stem cell marker, indicating that these cells arise from a small population within KLM1 after stress-induction by GEM (56). The action of metformin on cancer stem cells (CSCs), as reported for breast CSCs, appears to be mostly via depletion of nucleotide triphosphates (NTPs) rather than by exerting an effect on glycolytic and TCA cycle intermediates together with an inhibition of a signal transduction pathway that results in an inflammatory response (57, 58) dependent on nuclear factor-κB (NF-κB) and signal transducer and activator of transcription 3 (STAT3) (59, 60). The decreased NTP levels in CSCs reduce the availability of intermediates for energetics and biosynthesis of nucleic acids (DNA, RNA) and co-factors (e.g. flavin adenine dinucleotide (FAD), NADH, coenzyme A (CoA)) (42). This is corroborated in the current work in which it appears that CSCs in KLM1-R have a lower level of intracellular L-lactate (Figure 4B) compared to KLM1 (Figure 4A) in the presence of FBS.
Overall we corroborate the theory proposed by Scotland et al. (48) that metformin induces early inhibitory effects (peaking at 6h) on cell energetics via Akt signalling, which results in long-term (past 24 h) effects leading to inhibition of cell proliferation and apoptosis.
Our findings are, however, discordant with a previous report in pancreatic cancer (using 1 mM metformin for 16 h on PANC-1 and MiaPaCa-2), where metformin abolished mTORC1 activation without over-stimulating Akt phosphorylation on Ser-473 and prevented mitogen-stimulated ERK activation (61).
As a general note of caution, however, several recent publications agree that the metabolic effects of metformin may differ considerably between different cancer cell types and stage of cellular transformation, as well as undergo changes over time in response to cellular adaptation to the early inhibitory effects of metformin (42, 48, 61, 62). Our data, in addition, show that the micro-environment of the tumor also plays a role, as observed by the differences in energetic profiles in the presence or absence of FBS.
Acknowledgements
Western blot detection by Image Reader LAS-1000 Pro was carried-out at the Gene Research Centre of Yamaguchi University.
- Received December 31, 2014.
- Revision received January 20, 2015.
- Accepted January 22, 2015.
- Copyright© 2015 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}