


Abstract
Background/Aim: Ferroptosis is a recently identified form of regulated necrosis that can be experimentally induced in cancer cells with the chemical inducer erastin. Recently, we identified sorafenib, an inhibitor of oncogenic kinases, as an inducer of ferroptosis in hepatocellular carcinoma cells. Whether sorafenib is able to exert its ferroptotic activity in cancer cells originating from other tissues is presently unclear. Materials and Methods: We compared the levels of ferroptosis induced by sorafenib with those induced by the reference compound erastin in a panel of ten human cell lines originating from various tissues. Results: Sorafenib induced ferroptosis in different cancer cell lines. We found a positive correlation between the ferroptotic potency of sorafenib and erastin. Compared to other kinase inhibitors, sorafenib is the only drug that displays ferroptotic efficacy. Conclusion: The findings establish sorafenib as the first clinically-approved anticancer drug that can induce ferroptosis.
Induction of programmed cell death is an important goal of the medical treatment of cancer and apoptosis is often considered as the main form of programmed cell death induced by anticancer drugs (1, 2). Recently however, there has been a strong interest in non-apoptotic forms of cell death, such as autophagic cell death and regulated necroses (1, 2). Because cancer cells almost systematically resist apoptosis, these non-apoptotic forms of cell death are increasingly recognized as a promising field of research for the improvement of medical treatment of cancer (2).
Ferroptosis is a form of regulated necrosis that was first identified in 2012 in an experimental context by applying the chemical inhibitor erastin on cancer cells (3). Among regulated necroses, ferroptosis is characterized by the occurrence of oxidative stress and intense membrane lipid peroxidation leading to loss of selective permeability of plasma membrane (3-5). The name ferroptosis originates from the fact that iron chelation, by preventing the formation of reactive radical species such as the radical hydroxyl, protects cells from this form of cell death (3). The actual reference inducer of ferroptosis, erastin, was initially developed in order to target cancer cells bearing mutations of the RAS oncogene (6). Recently however, the study by Yang et al. addressing the impact of genomic alterations on ferroptosis induced by erastin in cancer cells found no correlation between the presence of RAS mutations and the cell's sensitivity to erastin (5). It is, therefore, unclear whether certain specific genomic alterations associated with human tumorigenesis promote ferroptosis. It is also unclear whether ferroptosis can be useful in the treatment of human tumours since the compounds reported to induce ferroptosis have not yet been approved for clinical use.
Recently, we identified sorafenib, an inhibitor of oncogenic kinases that is approved as an anticancer drug in the clinical setting, as an inducer of ferroptosis in hepatocellular carcinoma (HCC) cell lines (7). This conclusion was based on the observation that the cytotoxic effect of sorafenib on HCC cells was largely prevented by iron chelation by deferoxamine (7), but it nevertheless raised several questions. Is sorafenib only able to induce ferroptosis in HCC cells? Is sorafenib as potent as erastin at inducing ferroptosis? Is the molecular mode of action of sorafenib related to that of erastin? Is sorafenib active as an inducer of ferroptosis in cancer cells bearing defined alterations of the main oncogenic pathways? In the present study, we aimed to address these questions by comparing the effects of sorafenib with those of erastin in a panel of ten human cancer cell lines originating from various types of solid tumors.

Sorafenib induces ferroptosis in different human cancer cell lines. A. Phase-contrast pictures of the ACHN human kidney cancer cells exposed to sorafenib (10 μM) for 18 h. Ferroptotic cells, presenting a typical necrotic morphology, are indicated with black arrowheads. B. Cellular extracts prepared from ACHN cells exposed to sorafenib (10 μM) or doxorubicin+ABT-737 for 9h were analysed by immunoblotting for their expression of active Casp-3 or their substrate Poly-ADP-ribose-Polymerase (PARP). Deferoxamine (DFX, 200 μM) and the caspase inhibitor zVAD-fmk (50 μM) were applied in order to block ferroptosis and apoptosis, respectively. Actin labelling is shown as a loading control. C. LDH release in the ACHN exposed to sorafenib or erastin for 18 h. Cells were incubated with sorafenib (10 μM) or erastin (5 μM) and treated simultaneously with DFX and zVAD-fmk. LDH release is presented as a % of total LDH, and * indicates a p<0.05 difference compared to equivalent conditions with sorafenib alone. D. Correlation analysis between the % of ferroptosis induced by sorafenib (10 μM) and erastin (5 μM) in a panel of human cancer cells. Each dot represents a cell line. Cell lines that were found to be not sensitive to ferroptosis induced by sorafenib or erastin (lower left corner) are not annotated. The data presented are from 3 independent experiments. The Pearson's r value, as well as the associated p value, is indicated.
Materials and Methods
Cell lines. Huh7 and PLC/PRF5 cells (hepatocellular carcinoma) were obtained from Dr. Wychowski (Institut de Biologie de Lille, France) and were authenticated using profiling of short tandem repeats at 16 loci (LGC Standards, Molsheim, France). Caki-1 and ACHN (kidney tumours), SK-MEL-3 (melanoma), NCI-H460 (lung carcinoma), PANC-1 and BxPC-3 (pancreatic adenocarcinoma), and HCT116 and HT-29 (colon carcinoma) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Their mutational characteristics were extracted from the Cosmic database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/) (8). All cell lines were cultured in the recommended culture medium supplemented with 10% fetal calf serum (Jacques Boy, Reims, France), 2 mM glutamine and penicillin/streptomycin.
Immunoblots. Total extracts were prepared as described previously (9). Proteins were precipitated, loaded on SDS-PAGE and transferred to nitrocellulose membranes. Chemiluminescence was used for revelation.
Antibodies and reagents. Antibodies raised against Extracellular Regulated Kinase 1/2 (ERK1/2), ERK1/2 phosphorylated on Thr202/Tyr204 (pERK), active caspase-3 and cleaved Poly-ADP-Ribose Polymerase (PARP) were from Cell Signaling Technology (Danvers, MA, USA). Antibodies against β-actin were from Sigma (Saint-Quentin Fallavier France). Secondary antibodies coupled with horseradish-peroxidase were purchased from GE Healthcare (Aulnay sous bois, France). Sorafenib, erlotinib, gefitinib, tivantinib, selumetinib, imatinib, masatinib, vemurafenib and ponatinib were purchased from Euromedex (Souffelweyersheim, France). Erastin and rapamycin were purchased from Sigma.
LDH release assay. Lactate dehydrogenase (LDH) released in the culture medium was measured with the CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega Charbonnieres, France). The results were expressed as % of released LDH, taking complete cell extract as 100%.
Statistical analyses. The Student's t-test and Fisher's exact test were used and a value of p<0.05 was considered as threshold for significance. Correlation coefficients were calculated using the Pearson's test. Statistical analyses were performed with R3.02 (http://www.r-project.org/).
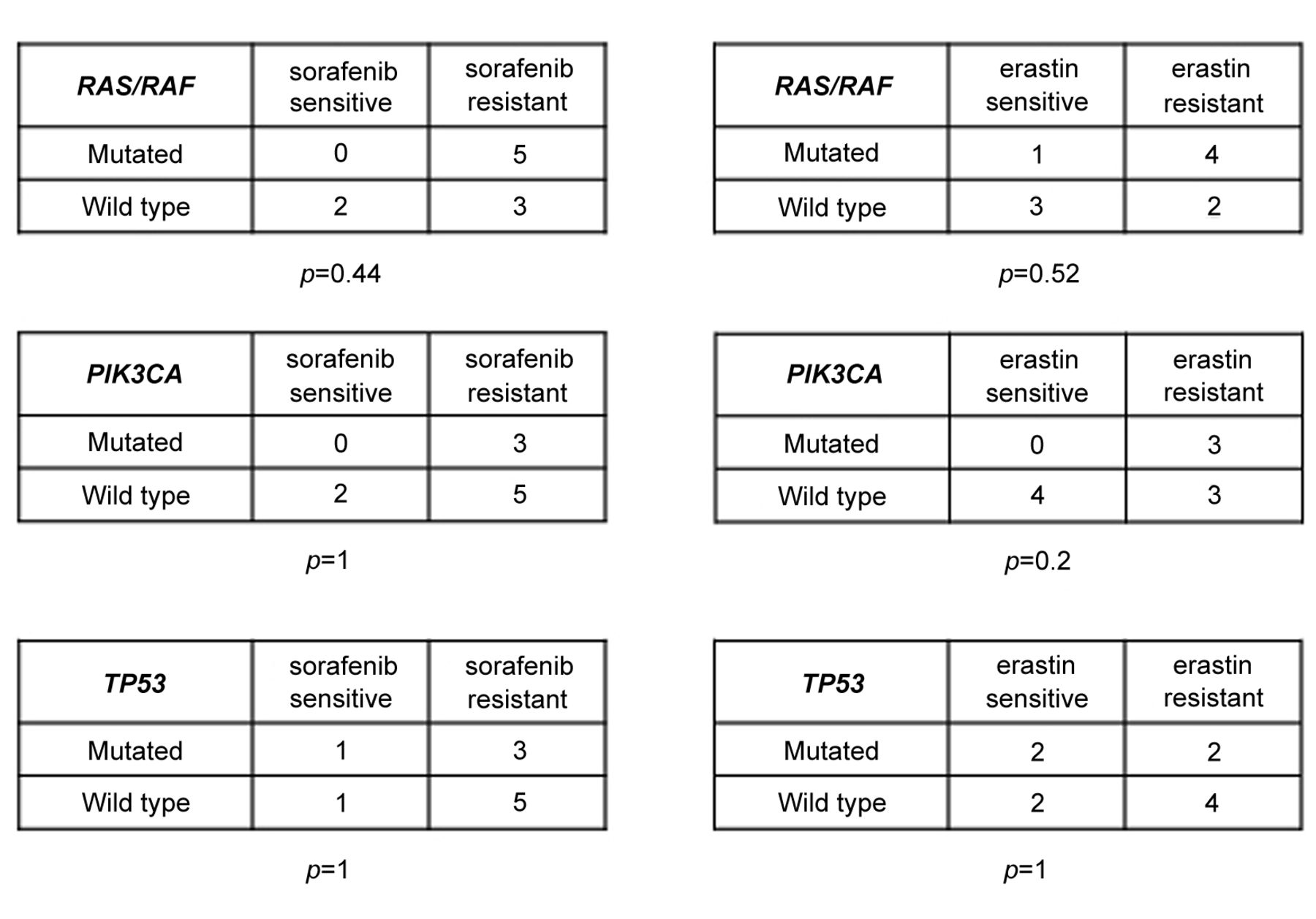
Sorafenib's ability to induce ferroptosis is independent of the presence of the most frequent oncogenic mutations. For each mutated gene, we present a contingency table with the number of cell lines found sensitive or resistant to ferroptosis induced by sorafenib or erastin. Ferroptosis-sensitive cells were arbitrarily defined by taking a value of 10% ferroptosis measured by LDH release as threshold. The p-values were calculated using the Fisher's exact test.
Results
Sorafenib induces ferroptosis in cancer cells originating from various tissues. In order to examine whether the induction of ferroptosis by sorafenib was restricted to hepatocellular carcinoma cells, we analyzed its effect in a panel of 10 human cell lines from different solid tumors. In addition to HCC cells (Huh7 and PLC/PRF5), sorafenib was applied on kidney cancer cells (Caki-1, ACHN), melanoma (SK-MEL-3), lung carcinoma (NCI-H460), pancreatic adenocarcinoma (PANC-1, BxPC-3) and colon carcinoma (HCT116, HT-29). We found that sorafenib exerted toxicity that was distinct from apoptosis in different human cell lines. For example, in the ACHN kidney cancer cell line, sorafenib induced cytotoxicity without the morphological manifestations of apoptosis (Figure 1A) or the activation of caspases (Figure 1B). Deferoxamine (DFX) almost completely protected ACHN cells from the cytotoxic effect of sorafenib, in contrast to the caspase inhibitor zVADfmk applied at concentrations found to prevent the activation of Caspase-3 induced by the chemotherapeutic compound doxorubicin in ACHN cells (Figure 1B). Using cell membrane permeability measurement with the LDH release assay, we found that sorafenib efficiently induced plasma membrane lysis of ACHN cells that could almost totally be prevented by the application of DFX (Figure 1C). The same pharmacological profile was found with erastin, the reference inducer of ferroptosis (Figure 1C). We concluded that sorafenib is able to induce ferroptosis in cancer cells originating from different tissues and not only in HCC.
In order to measure the extent of ferroptosis induced in every cell line in our panel, a % rate of ferroptosis was calculated by subtracting LDH release after iron chelation from the values measured upon exposure to sorafenib (10 μM) or erastin (5 μM) for 18 h (Figure 1D). As shown in Figure 1D, we found that the cell lines presented great variability in terms of sensitivity to the induction of ferroptosis by sorafenib and erastin. However, a positive correlation was found between the % ferroptosis induced by sorafenib and that produced by erastin. Interestingly, in some cases, sorafenib or erastin were able to induce ferroptosis with different efficacies. For example, in PLC/PRF5 cells, sorafenib induced close to 10% ferroptosis but erastin was inefficient. Reciprocally, Caki-1 and PANC-1 cells were highly sensitive to ferroptosis induction by erastin, but sorafenib was poorly-efficient in these cells (Figure 1D). Therefore, human cancer cells display heterogeneous susceptibility to ferroptosis induced by sorafenib.

Dose-response effect of sorafenib on ferroptosis and ERK phosphorylation. A. Increasing concentrations of sorafenib and erastin (both 0-20 μM) were applied on ACHN for 18 h and the % of ferroptosis was calculated. B. the activation level of the RAF-MEK-ERK kinase cascade was analyzed by immunoblotting in ACHN cells exposed to the same conditions.
Sorafenib induces ferroptosis independently of the oncogenic status of cancer cells. In order to examine whether the oncogenic status of cancer cells modulates their susceptibility to ferroptosis, we attempted to correlate the % of ferroptosis with the mutational status of these cells. The mutational status of the RAS, RAF, PIK3CA and TP53 genes were taken into account, since these genes represent some of the most important mutational targets in human tumours (10, 11). The % of cells carrying mutations of the RAS, RAF, PIK3CA or TP53 oncogenes was compared between the two groups of cells, considered sensitive and resistant to sorafenib and erastin, respectively (taking a value of 10% ferroptosis measured by LDH release as threshold to identify ferroptosis-sensitive cells). No statistically significant difference in the frequency of mutations was found (Figure 2), suggesting that these mutations were not associated with a heightened sensitivity to ferroptosis induced by erastin or sorafenib.

Ferroptosis induction is a specific property of sorafenib. ACHN cells were exposed to sorafenib (10 μM), erlotinib (1 μM), gefitinib (1 μM), tivantinib (1 μM), vemurafenib (10 μM), selumetinib (1 μM), rapamycin (1 μM), imatinib (10 μM), masatinib (1 μM) or ponatinib (1 μM). Results are expressed as % of ferroptosis, calculated by subtracting LDH released in the presence of DFX (200 μM) from LDH released in the culture medium of cells exposed to the indicated inhibitors for 18 h. *p<0.05 difference compared to control condition.
Ferroptosis induction is a property that is unique to sorafenib compared to other kinase inhibitors. Sorafenib is a kinase inhibitor with a relatively weak specificity, but the RAF kinases (essentially BRAF and CRAF) are presumed to be essential targets of sorafenib (12). In ACHN cells, sorafenib was found to exert a significant ferroptotic activity at concentrations above 10 μM (Figure 3A). Under these conditions, sorafenib did not prevent the activation of the RAF-MEK-ERK cascade (Figure 3B).
Induction of ferroptosis could either be a general property of kinase inhibitors or a singular property of sorafenib. In order to address this question, we applied 10 clinically-relevant kinase inhibitors, presenting with different chemical reactivities on the kinome of cancer cells (13): erlotinib, gefitinib, tivantinib, vemurafenib, selumetinib, rapamycin, imatinib, masatinib and ponatinib were applied on ACHN cells in culture (Figure 4). Although ACHN cells are highly sensitive to ferroptosis induced by sorafenib, none of the other kinase inhibitors were able to induce ferroptosis (Figure 4). We concluded that the induction of ferroptosis is a specific property of sorafenib and one that is not shared with other clinically-available anticancer drugs.
Discussion
While research on anticancer drugs has until recently been centered on their ability to induce apoptosis, other forms of cell death are emerging as effective means to eliminate cancer cells (1, 2). Ferroptosis is a recently described form of regulated necrosis that offers interesting perspectives in this respect (3-7). The induction of ferroptosis might be an interesting goal for the medical treatment of cancer, provided that drugs presenting a good efficacy and safety profiles are available in humans. In the present report, we show that sorafenib is able to induce ferroptosis in cancer cells originating from different tissues, such as the liver or the kidney. Sorafenib is therefore the first clinically-approved anticancer drug identified as an inducer of ferroptosis.
An important conclusion of our work is the fact that the induction of ferroptosis is unrelated to the RAF kinase-inhibitory effect of sorafenib. Sorafenib induced ferroptosis in cancer cells in which it did not efficiently block the RAF-MEK-ERK kinase cascade. In addition, we tested multiple kinase inhibitors that share overlapping reactivities with sorafenib but none displayed ferroptotic efficacy. While the ferroptotic mode of action of sorafenib remains unknown, we found a positive correlation between the levels of ferroptosis induced by sorafenib and those induced by erastin. This suggests that the two compounds exert a similar mode of action in their induction of ferroptosis. In agreement with this possibility, a recent report by Dixon et al. showed that sorafenib is able to block the cystine-glutamate exchanger Xc- and, therefore, the synthesis of glutathione (GSH) in a similar way as erastin (14). Future investigations centered on this aspect of cancer cellular physiology will be important to better-explain the ferroptotic activity of sorafenib in human cancer cells.
Finally, an interesting point that was observed in the present study is the high cell line-to-cell line variability in the susceptibility to ferroptosis. It is presently unknown what parameters, genetic or non-genetic, account for this heterogeneous susceptibility. In our panel of cancer cells we found that none of the genomic alterations most commonly found in human tumours were associated with increased sensitivity to ferroptosis induced by sorafenib or erastin. Based on the high number of parameters that regulate the redox metabolism of cancer cells (15-19), we speculate that ferroptosis might be under complex regulation in cancerous cellular states. Comprehensive studies and system biology approaches might, therefore, be required to explore the regulation of ferroptosis (20, 21). Because sorafenib is the only clinically-approved anticancer drug demonstrated to act as an inducer of ferroptosis, we believe that this drug will be instrumental for the understanding of ferroptosis regulation in humans.
Acknowledgements
We are grateful for the financial help received from the Ligue contre le Cancer - Comité de la Somme.
Footnotes
-
Conflicts of Interest
None to report.
- Received July 23, 2014.
- Revision received August 1, 2014.
- Accepted August 5, 2014.
- Copyright© 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- CCT3 drives Sorafenib resistance by inhibiting TFRC-mediated iron uptake in HCC
- HSPB1 facilitates chemoresistance through inhibiting ferroptotic cancer cell death and regulating NF-{kappa}B signaling pathway in breast cancer
- GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis
- MDM2 and MDMX promote ferroptosis by PPAR{alpha}-mediated lipid remodeling
- Drugs Repurposed as Antiferroptosis Agents Suppress Organ Damage, Including AKI, by Functioning as Lipid Peroxyl Radical Scavengers
- The crosstalk between autophagy and ferroptosis: what can we learn to target drug resistance in cancer?
- Ferroptosis-like signaling facilitates a potent innate defense against Plasmodium infection
- Regulation of lipid peroxidation and ferroptosis in diverse species