


Abstract
Background: Knowledge regarding substances that attenuate motility of cancer cells has gathered significant attention, as they benefit the development of novel anticancer strategies. The anti-migration and anti-invasion activities of artonin E, extracted from bark of Artocarpus gomezianus, were investigated in lung cancer cells in this study. Materials and Methods: Cytotoxicity and antiproliferative effects of artonin E were examined by 3- (4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Migration and invasion assays were performed on H460, H23, A549 and H292 human lung cancer cells. Cell morphology was determined by phalloidin-rhodamine staining. Motility-related proteins were investigated by western blotting. Results: Artonin E exhibited anti-migration and anti-invasion activities in H460 cells. Cell morphology revealed that treatment of the cells with non-toxic concentrations of artonin E resulted in a decrease of activated focal adhesion kinase (FAK), downstream protein kinase B (AKT) activation, and Cell division cycle-42 (CDC42), all of which were associated with the anti-motility effect of this compound. Artonin E inhibited invasion and migration of other lung cancer cells, namely H292, H23 and A549 cells. Conclusion: These results suggest that artonin E may be a promising candidate for anti-metastasis use.
Metastatic lung cancer is the cause of more than 90% lung cancer-related deaths worldwide (1). Although the earliest stage of the disease presents as only pulmonary nodule without lymph node spread, some patients with disease at this stage will finally die from undetectable metastases (2). Cancer metastasis is a complex process of cell spreading which can be divided into several steps including migration, invasion, intravasation, survival in the circulation, extravasation, and metastatic colonization (3, 4). A growing body of evidence suggests that migration and invasion are crucial steps for successful metastasis (5); however, at present, there are no approved drugs that inhibit such cancer cell behavior.
Even though the molecular mechanisms, which cancer cells use for migration and invasion are not fully understood, based on previous research, they involve the ability of cancer cells to change their affinity for the extracellular matrix (ECM) and such alterations are due to modifications of various cellular signaling pathways including focal adhesion kinase FAK (6). Indeed, the activation of FAK through phosphorylation is important for FAK-induced focal adhesion turnover and cell movement (7). Activated FAK can transduce the signal through the phosphorylation of protein kinase B (AKT) resulting in cellular responses such as cell invasion and migration (6). Recently, the Rho family of small guanosine-5’-triphosphatases (GTPases), especially Cell division cycle-42 (CDC42), were shown to play an essential role in modulating actin re-organization and filopodia formation (8) and its overexpression was shown to be associated with enhanced migration and cancer aggressiveness (9, 10). The expression level of CDC42 was found to be up-regulated in many types of cancer (10, 11). In lung cancer, CDC42 was shown to be highly overexpressed in primary lung cancer cells (12). A previous study indicated that both curcumin-mediated CDC42 down-regulation and CDC42 knock-down attenuated cancer cell invasion in vivo (12).
Artonin E (Figure 1), an active flavonoid, obtained from the bark of Artocarpus gomezianus Wall. exTréc. (Moraceae), is known as “Ka-Noon-Pah” in Thailand (13). Artonin E was shown to exhibit promising growth-inhibition action against breast cancer cells (14). However, the effects of artonin E on cancer cell migration and invasion are unknown. In our view, the knowledge regarding such activities of the compound would benefit the development of novel anti-metastasis drugs, as well as strategies to overcome cancer.
Materials and Methods
Test compound. Artonin E, a pure compound in dry powder form, was obtained from Associate Professor Boonchoo Sritularak, Departments of Pharmacognosy and Pharmaceutical Botany, Faculty of Pharmaceutical Sciences, Chulalongkorn University (BKK, Thailand), and was dissolved in ethanol and RPMI-1640 to achieve the desired concentrations, containing less than 0.1% ethanol at final dilution.
Cell culture. Human lung cancer H460, H292, H23 and A549 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were cultivated in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 100 units/ml penicillin/streptomycin in a 5% CO2 environment at 37°C.
Chemicals. Hoechst 33342, propidium iodide (PI), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and dimethyl sulfoxide (DMSO) were obtained from Sigma Chemical, Inc. (St. Louis, MO, USA). Rabbit antibodies to CDC42, caveolin-1 (CAV1), pFAK (Tyr 397), FAK, pAKT (Ser 473), AKT, and β-actin were obtained from Cell Signaling Technology, Inc. (Danver, MA, USA).
Cytotoxicity and cell proliferation assay. To determine artonin E-mediated cytotoxicity and effects on cell proliferation, cell viability was determined by the MTT assay, as previously described (15), which measures cellular capacity to reduce MTT (yellow) to purple formazan crystal by mitochondrial dehydrogenase. Cells were seeded in a 96-well plate and allowed to attach for 12 h. Cells were treated with different concentrations of artonin E (0-50 μg/ml) and incubated for 24 h for cytotoxicity assay and 12, 24, 48 and 72 h for proliferation assay, and cells were incubated with 100 μl of 500 μg/ml MTT solution for 4 h at 37°C. Then, MTT solution was removed and 100 μl of 99.9% DMSO was added to dissolve the formazan crystal. The intensity of formazan product was measured at 570 nm using a microplate reader. All analyses were performed in at least three independent replicate cultures. The percentage of cell viability was calculated as follows:

Apoptosis and necrosis assay. Apoptotic and necrotic cell death were determined by Hoechst 33342 and PI co-staining as described elsewhere (15). Cells were seeded in 96-well plates and treated with different concentrations (0-0.5 μg/ml) of artonin E for 24 h, cells were incubated with 10 μM of the Hoechst 33342 and 5 μg/ml PI dye for 30 min at 37°C. The apoptotic cells with condensed chromatin and/or fragmented nuclei and PI-positive necrotic cells were visualized under a fluorescence microscope (Olympus IX51 with DP70; Olympus, Center Valley, PA, USA).
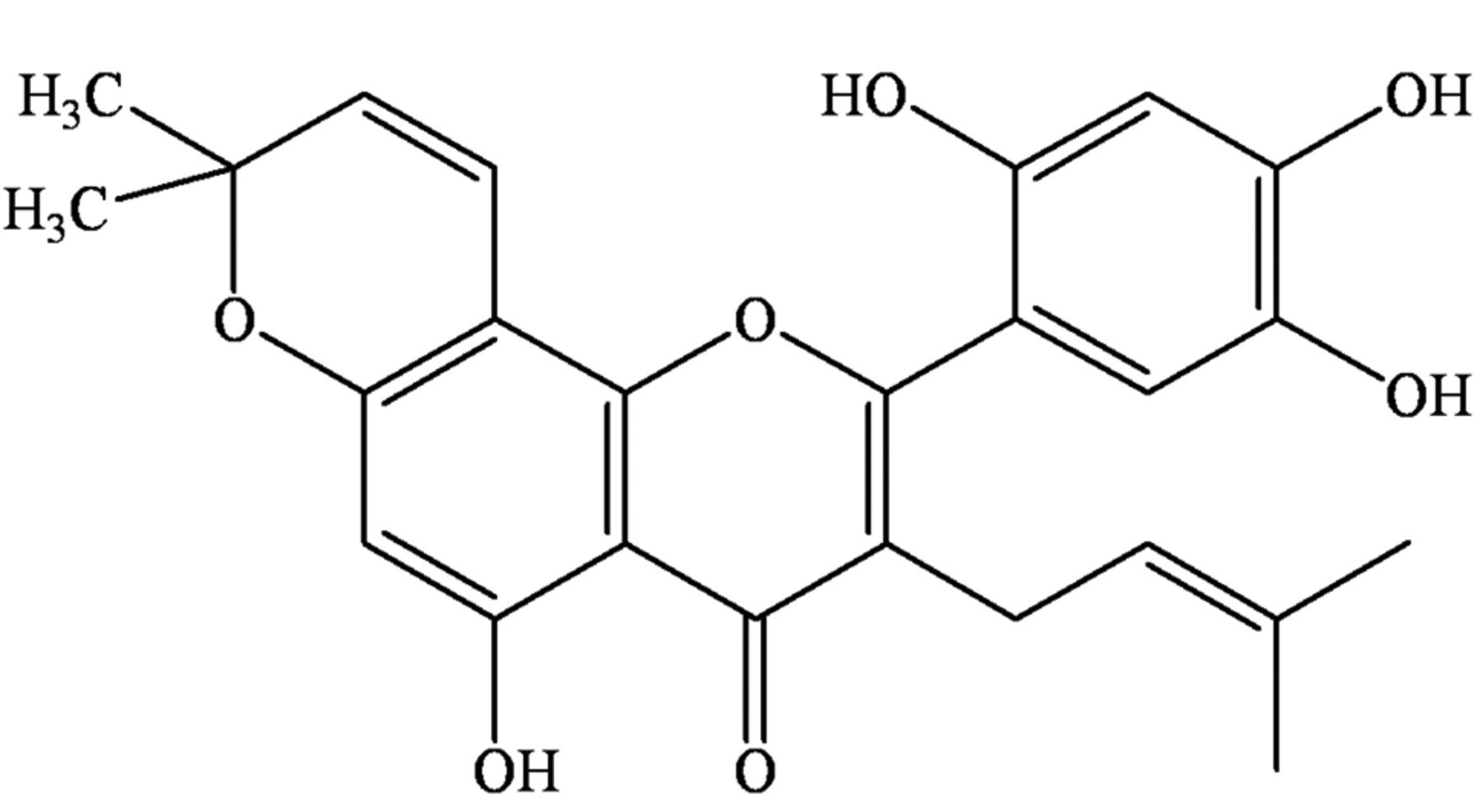
Structure of artonin E.
Wound-healing assay. The wound-healing assay was determined as previously described (16). Briefly, a monolayer of cells was cultured in 96-well plates, and then a wound space was created by a 1-mm width tip. After rinsing with PBS, the cell monolayers were treated with different concentrations of artonin E (0-0.5 μg/ml) and allowed to migrate for 24, 48, and 72 h. Micrographs were taken under a phase-contrast microscope (×100; Olympus IX51 with DP70), and wound spaces were measured from 10 random fields of view using Olympus DP controller software. Quantitative analysis of cell migration was performed by using an average wound space from random fields of view, and the relative migration was calculated.
Invasion assay. An invasion assay was conducted using a 24-well Transwell unit with polyvinylidene difluoride (PVDF) filters (8 μm pore size) as previously described (16). Each well was pre-coated with 50 μl Matrigel (BD Biosciences, Bedford, MA, USA). The lower chamber of the unit contained with RPMI medium containing 10% FBS. Cells at the density of 3×105 cells/100 μl were incubated with artonin E at different concentrations (0-0.5 μg/ml) in RPMI containing 1% FBS, and then added to the upper chamber. After 24 h, the top medium and Matrigel were completely removed and the bottom side was fixed with 3.7% paraformaldehyde. After staining cells with Hoechst 33342, cells were visualized and scored under a fluorescence microscope (Olympus IX51 with DP70).
Cell morphological characteristics. Cells were washed with phosphate buffer saline (PBS) and fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. Afterwards, cells were permeabilized by 0.1% Triton-X100 in PBS for 4 min, washed with PBS three times, and blocked with 0.2% bovine serum albumin (BSA) for 30 min as described elsewhere (17). After washing, cells were incubated with rhodamine-phalloidin in PBS for 30 min and rinsed three times. Images of stained cells with rhodamine-labeled phalloidin were taken under a fluorescence microscope (Olympus IX51 with DP70).
Western blot. Cells were seeded in 6 well-plates and treated with different concentrations of artonin E (0-0.5 μg/ml) for 24 and 72 h. After specific treatments, cell lysates were obtained by incubating the cells in ice-cold lysis buffer containing 20 mM Tris-HCl (pH 7.5), 0.5% Triton X-100, 150 mM sodium chloride, 10% glycerol, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 100 mM phenylmethylsulfonyl fluoride, and a protease inhibitor cocktail (Roche Molecular Biochemicals, Indianapolis, IN, USA) for 60 min on ice. Protein content was determined using the Bradford method (Bio-Rad Laboratories, Hercules, CA, USA) and an equal amount of protein from each sample (60-80 μg) was heated at 95°C for 5 min with Laemmli loading buffer. The lysates were then loaded onto 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel for electrophoresis. After separation, proteins were transferred onto 0.45 μm nitrocellulose membranes (Bio-Rad). The transferred membranes were blocked for 1 h in 5% non-fat dry milk in TBST [25 mM Tris-HCl (pH 7.5), 125 mM NaCl, 0.05% Tween 20]. Membranes were washed twice with TBST for 7 min and incubated with the primary antibodies at 4°C for 10 h. Membranes were then washed three times with TBST for 7 min and incubated with horseradish peroxidase-coupled isotype-specific secondary antibodies for 2 h at room temperature. After washing again, the immune complexes were detected by enhancing with chemiluminescence substrate (Supersignal West Pico; Pierce, Rockford, IL, USA), and quantified using analyst/PC densitometry software (Bio-Rad) normalized to the level of β-actin protein.
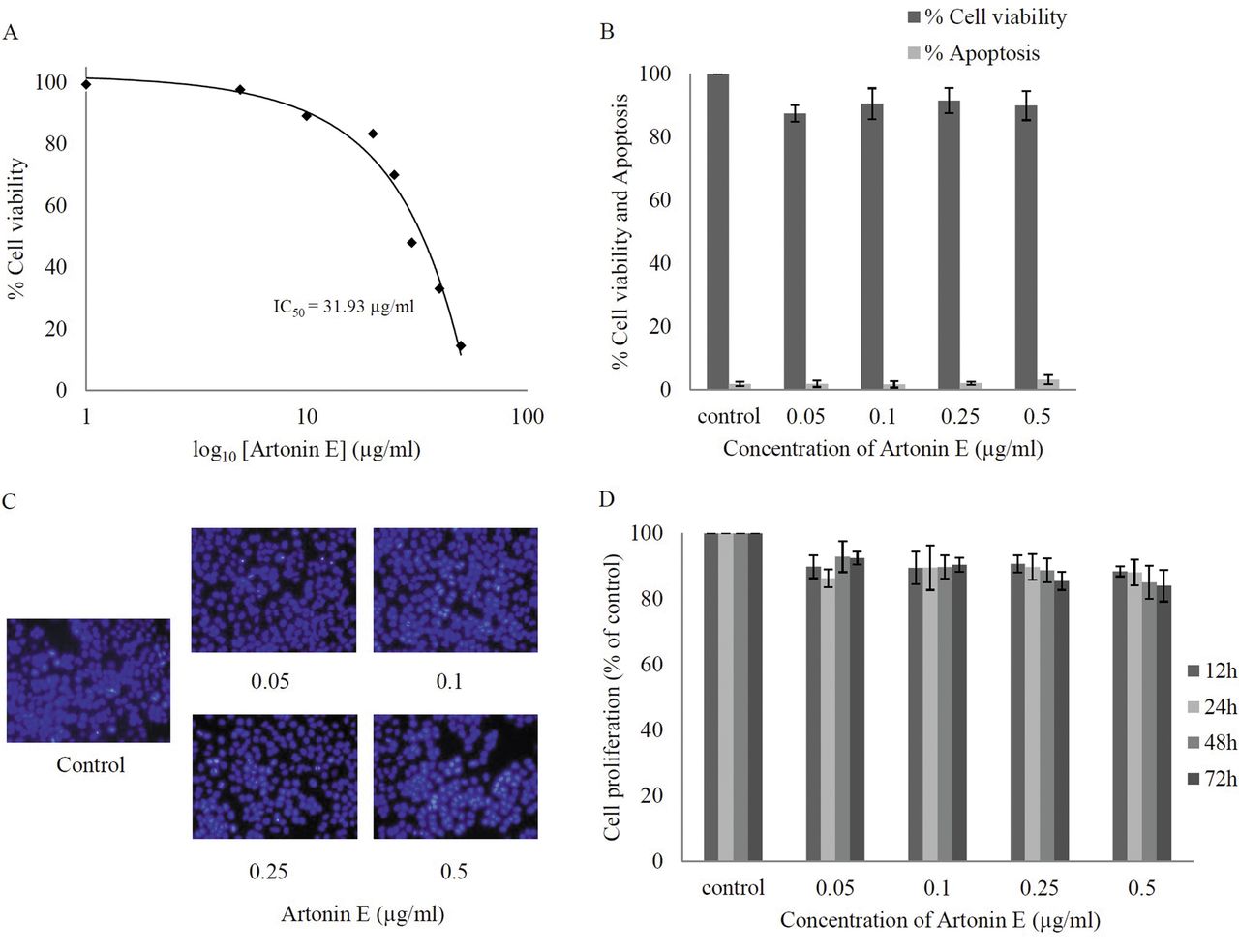
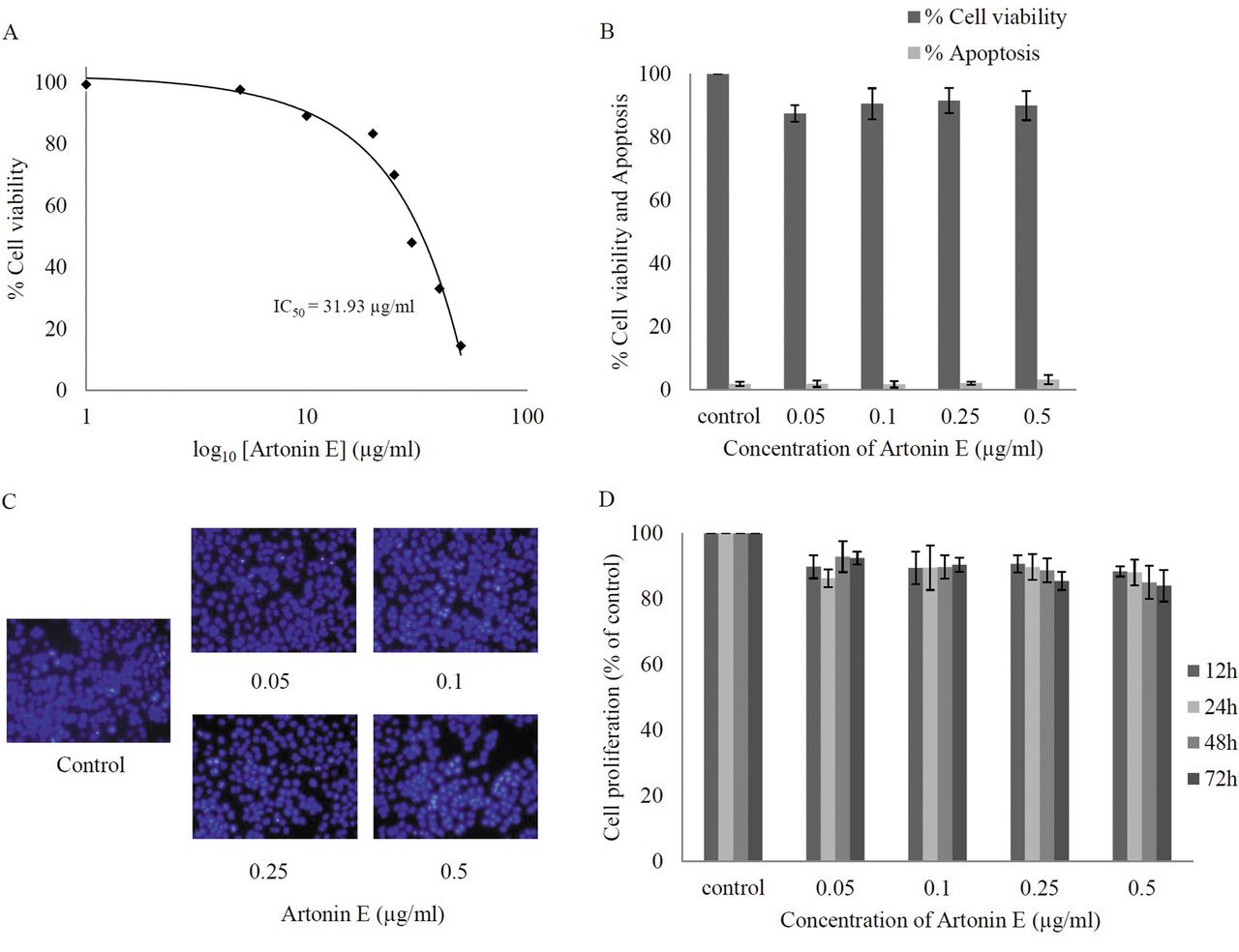
Effect of artonin E on cell viability of human lung cancer H460 cells. Cells were treated with different concentrations of artonin E (0-50 μg/ml) for 24 h. A: Cytotoxicity was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and the concentration for 50% cell survival (IC50) was determined. B: Percentage of cell viability and cell apoptosis were analyzed by MTT assay and Hoechst 33342 staining assays, respectively. C: Morphology of apoptotic nuclei stained with Hoechst 33342 and propidium iodide (PI). D: Proliferation of H460 cells in response to artonin E 0.05-0.5 μg/ml for 12, 24, 48, and 72 h was investigated by the MTT assay. Values are means of triplicate samples±SD.
Statistical analysis. Mean data from at least three independent experiments were normalized to the result of the untreated control. Statistical differences between means were determined using an analysis of variance (ANOVA) and post hoc test at a significance level of p<0.05, and data are presented as the mean±SD.
Results
Cytotoxic effect of artonin E on human non-small cell lung cancer H460 cells. Although the anticancer activity of artonin E has been previously demonstrated in a breast cancer cell model (14), it is not clear whether such a compound causes cytotoxicity to lung cancer cells. The present study first-characterized the cytotoxic effect of artonin E on H460 human lung cancer cells by incubating the cells in the presence and absence of artonin E (0-50 μg/ml) for 24 h, and cell viability was analyzed by the MTT assay. Figure 2A shows that treatment with artonin E caused a dose-dependent decrease in cell survival and 50% inhibition (IC50) was observed in response to artonin E at the concentration of 31.93 μg/ml. Because the study aimed to investigate antimigration and anti-invasion activities of the compound, the concentrations of artonin E which cause neither toxic nor proliferative effects were further clarified. Results indicated that treatment with artonin E at concentrations of 0.05-0.5 μg/ml caused no significant effect on H460 cell viability (Figure 2B). The nuclear morphology study shown in Figure 2C supported the above finding that no apoptotic or necrotic cell death was detected in response to artonin E at these concentrations. In addition, artonin E at concentrations of 0.05-0.5 μg/ml did not significantly alter proliferation of the cells up to 72 h (Figure 2D).
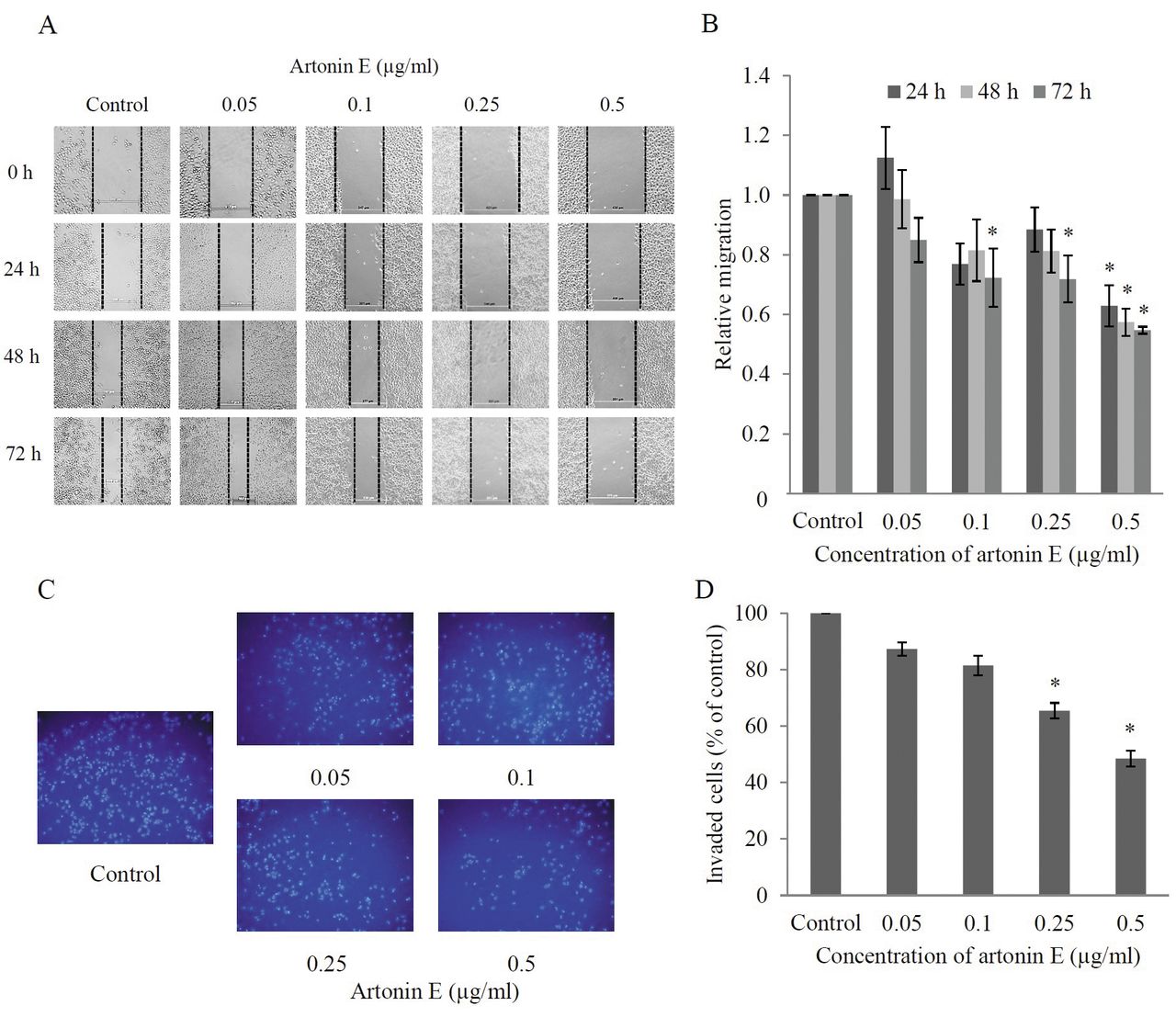
Effects of artonin E on H460 cell migration and invasion. For migration assay, wound-healing assay was performed. Wound space was created and the cells were treated with sub-toxic concentrations of artonin E for different times. For invasion assays, cells were seeded onto Matrigel-coated membrane and treated with sub-toxic concentrations of artonin E for 24 h. A: Wound space was visualized under a phase-contrast microscope at the indicated times. B: The relative cell migration was analyzed by comparison of the relative change in wound space of the treated groups over that of the untreated control. C, D: The invaded cells were stained with Hoechst 33342, visualized under fluorescence microscopy, and the relative cell invasion was determined. Values are means of triplicate samples±SD.; *p<0.05 versus untreated control.
Artonin E inhibits migration and invasion of H460 cells. To examine the effect of artonin E on migration of the cells, a wound healing assay was performed. Briefly, the confluent monolayer of H460 cells was scratched and cells were cultured with or without sub-toxic concentrations of artonin E (0.05-0.5 μg/ml) for 24, 48, and 72 h. Figures 3A and B show that the incubation of cells with artonin E at a concentration of 0.5 μg/ml significantly reduced the spreading of H460 cells to the wound area as early as 24 h, whereas artonin E at 0.05 μg/ml had no significant effect on cell migration in comparison to that of the untreated controls. In addition, artonin E at concentrations of 0.1 and 0.25 μg/ml significantly inhibited migration of H460 cells at 72 h. These results indicate that artonin E possesses the ability to inhibit cancer cell migration.
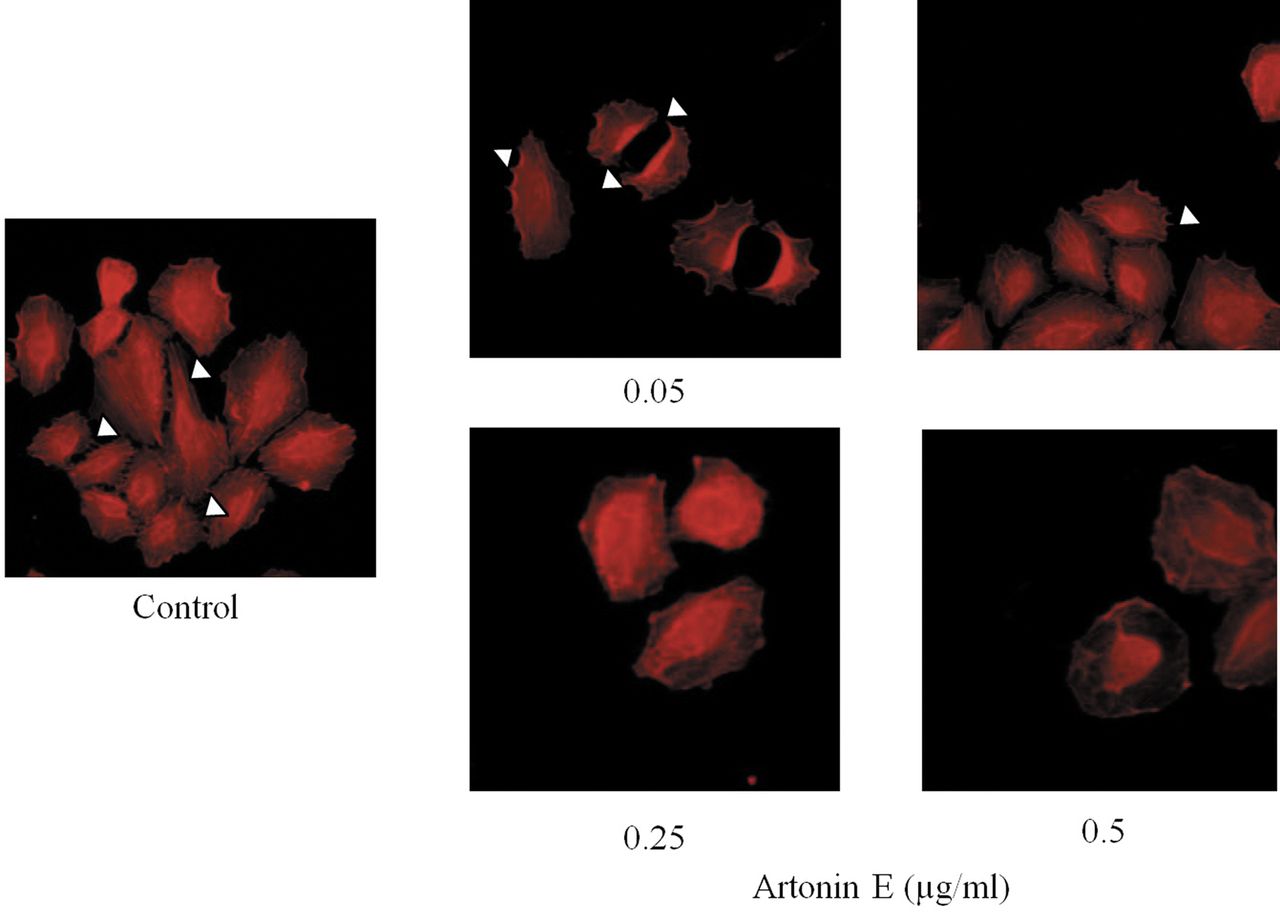
Effect of artonin E on filopodia alteration. After treating with sub-toxic concentrations of artonin E for 24 h, cells were stained with phalloidin and filopodia were examined under fluorescent microscopy. Filopodia are indicated by arrow.
For invasion assays, the cancer cells were added to a 24-well transwell pre-coated with Matrigel and treated with different concentrations of artonin E. The invaded cells were determined as described in Materials and Methods. As shown in Figures 3C and D, artonin E at 0.25 and 0.5 μg/ml significantly inhibited cancer cell invasion through Matrigel matrix at 24 h.
Artonin E inhibits filopodia formation in lung cancer cells. During cell movement, membrane protrusions called filopodia were shown to be increased and the formation of filopodia was shown to be closely involved with cancer cell migration and invasion (18). Having shown that artonin E at sub-toxic concentrations significant inhibited lung cancer cell migration and invasion, we further tested whether the compound has an effect on filopodia formation. Cells were treated with 0-0.5 artonin E and phalloidin-labeled filopodia were detected under fluorescence microscopy. Figure 4 shows that in untreated controls, cells exhibited several membrane protrusions of filopodia. Interestingly, treatment with artonin E dramatically reduced directional stress fibers and filopodia in H460 cells in comparison to that of untreated cells. These data and the above findings suggest that artonin E has a negative impact on cell migration and invasion and this may, at least in part, involve the reduction of cellular filopodia.
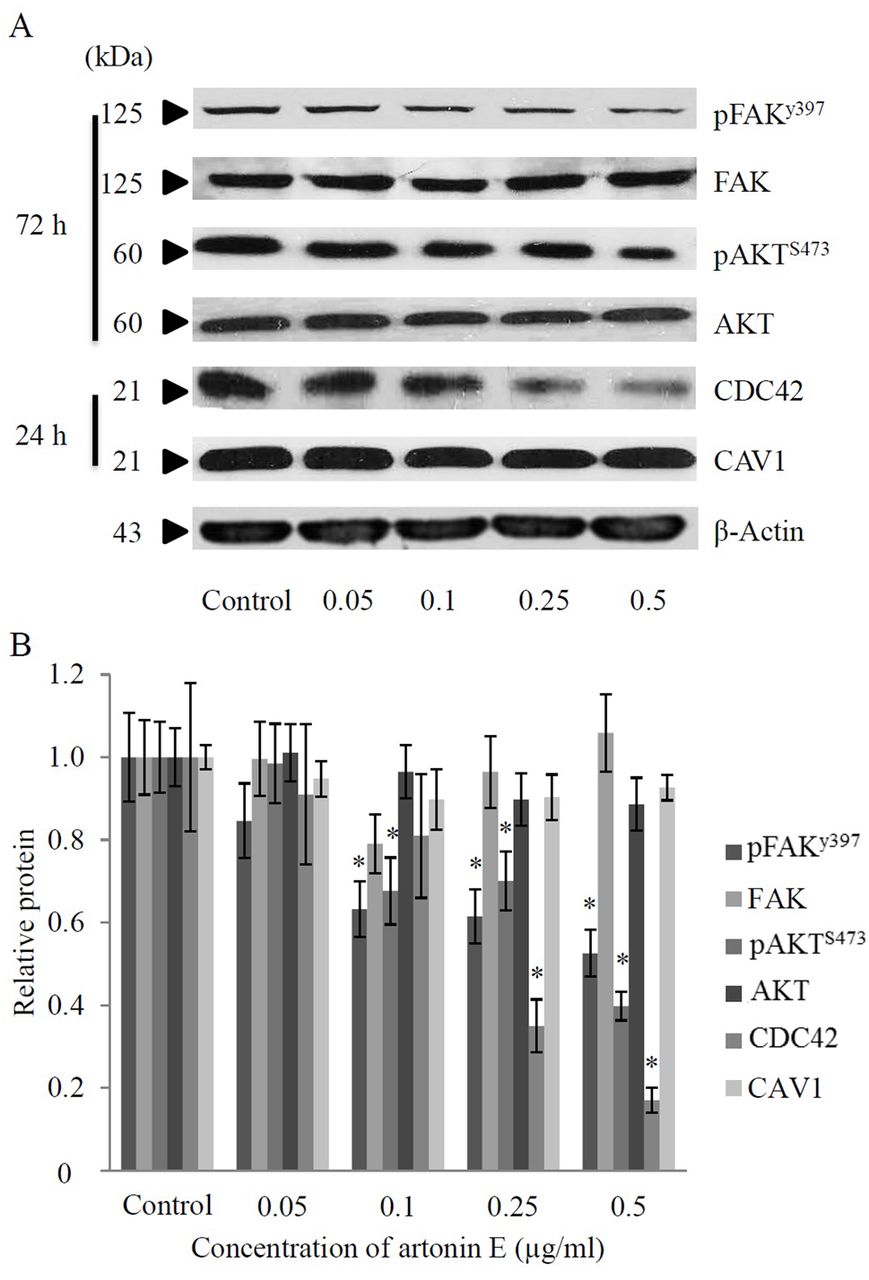
Artonin E inhibits FAK signaling and suppresses CDC42 expression in H460 cells. In order to clarify the mechanism of artonin E in suppression of cancer cell motility, the expression level and activation status of proteins regulating cell motility, namely FAK and AKT, were investigated. Cells were seeded and incubated in the presence or absence of artonin E (0.05-0.5 μg/ml) and the expression of proteins was determined by western blotting. While artonin E had only a minimal effect on the level of total FAK, treatment of artonin E at concentrations of 0.1-0.5 μg/ml significantly reduced the level of phosphorylated FAK at Tyr 397 (activated FAK). It has been well-established that the phosphorylation of FAK at Tyr 397 activates its kinase activity that further triggers AKT signaling (6). We, thus, investigated the possible effect of artonin E on AKT activation. Figure 5 demonstrates that artonin E significantly reduced phosphorylation of AKT at Ser 473 in a concentration-dependent manner while it had no significant effect on total AKT expression. These findings suggested that artonin E inhibits cell migration via an FAK-AKT-dependent mechanism.

Effect of artonin E on focal adhesion kinase (FAK), protein kinase B (AKT), Cell division cycle (CDC42), and caveolin-1(CAV1) proteins. A: Cells were seeded and treated with different concentrations of artonin E (0-0.5 μg/ml) for 24 and 72 h and then the expressions of pFAK (Tyr 397), FAK, pAKT (Ser 473), AKT, CDC42 and CAV1 proteins were determined by Western blotting. To confirm equal loading of samples, blots were re-probed with β-actin antibody. B: The immunoblot signals were quantified by densitometry and mean data from four independent experiments were presented. Values are means of samples±SD. (n=4); *p<0.05 versus untreated group.
The high expression level of CDC42, as well as CAV1 was shown to be tightly-associated with an increase in aggressiveness of cancer (9, 19). Since the CDC42 was shown to be up-regulated in primary lung cancer cells and such an increase of the protein was shown to be associated with high TNM stage and lymph node metastasis, it is interesting to investigate whether artonin E treatment could affect the cellular CDC42 level. We assessed the effect of artonin E on CDC42 level by western blot analysis and found that CDC42 protein expression was down-regulated in response to artonin treatment at the concentrations of 0.25 and 0.5 μg/ml in comparison to that of the untreated control. However, the expression of CAV1 protein was found to be only slightly affected in response to artonin E treatment. These results suggest that artonin E attenuates lung cancer cell migration and invasion through an FAK-AKT-dependent mechanism and via reduction of CDC42.
Artonin E inhibits migration and invasion of other lung cancer cell lines. In order to confirm the negative regulatory effect of artonin E on lung cancer cell migration and invasion, human lung cancer cell lines H292, H23 and A549 were treated with the non-toxic concentrations of artonin E and subjected to migration as well as invasion assays. Our results, shown in Figure 6, revealed that artonin E at 0.25 and 0.5 μg/ml significantly inhibited the migratory behavior of H292, H23, and A549 cells in comparison to that of the respective untreated controls. Consistently, the invasion assay indicated that artonin E at such concentrations significantly suppressed the invasion of H292, H23, and A549 cells (Figure 7). These data strengthen the observations of the present study that artonin E has the ability to inhibit migration and invasion of lung cancer cells.
Discussion
Advanced and novel strategies for cancer therapies, including those inhibiting metastasis of cancer, have gathered increasing attention. Limited efficacy has been obtained from the available therapy, resulting in only 68% 5-year survival of patients with cancer in the United States (20), and the major cause of death found in such patients involves metastasis. As a hallmark of cancer metastasis, the ability of cancer cells to migrate away from the original tumor and to invade the blood or lymphatic circulation is considerably important (3). Herein we demonstrated, to our knowledge for the first time that artonin E, a plant-derived pure compound has a promising ability to inhibit lung cancer cell movement. Activation of cancer cell motility involves several mechanistic pathways including FAK (6), AKT (21), CDC42 (22), and CAV1 (19). Tumor progression and metastasis can be stimulated by FAK signaling pathways through the regulation of cell migration and invasion. Mutagenesis of FAK gene at the 397 site by the insertion technique eliminated the ability of FAK to activate motility (23). Likewise, many studies demonstrate that phosphorylation of FAK at Tyr 397 is necessary for FAK to promote cell migration (24-26). Invasion of malignant cells is also promoted by FAK signaling (27). Moreover, evidence has indicated that FAK triggers and activates the Phosphoinositide 3-kinase (PI3K)-AKT pathway (6). AKT activation was shown to be associated with cell migration and invasion by several means (21, 28). For instance, AKT regulates the stability of microtubules, which has an essential role in cell motility (29) and its function was shown to increase invasiveness of cells (21).

Effects of artonin E on migration of H292, H23, and A549 cells. H292, H23, and A549 cells were subjected to migration assay and visualized under a phase-contrast microscope. The relative cell migration of H292, H23, and A549 cells are shown in A, B, and C, respectively. Values are means of triplicate samples±SD.; *p<0.05 versus untreated control.
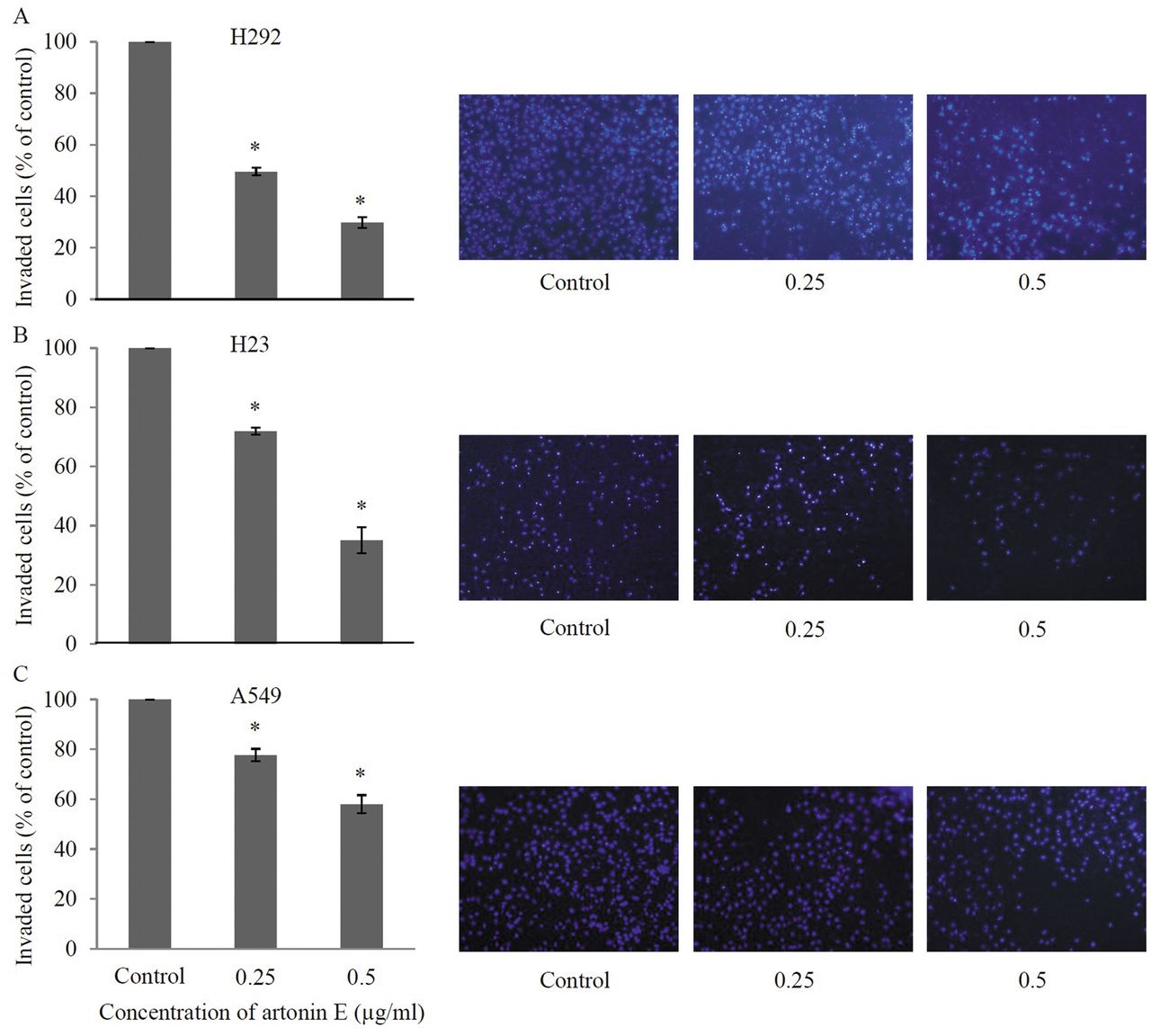
Effects of artonin E on invasion of H292, H23, and A549 cells. H292, H23, and A549 cells were added to Matrigel and treated with sub-toxic concentrations of artonin E for 24 h. The invaded cells were stained by Hoechst 33342 and visualized under fluorescence microscopy. Values are means of triplicate samples±SD.; *p<0.05 versus untreated control.
CDC42 protein, a member of Rho GTPase family, has been shown to regulate many cellular processes, including actin re-organization and cell polarity (30). CDC42 is also implicated in filopodia formation which leads to migration and invasion of cancer cells (31). Some evidence indicate that CDC42 was overexpressed in many types of human carcinoma, resulting in increased aggressiveness of the cancer (9, 11). Furthermore, the knock-down of CDC42 generally resulted in inhibition of cancer cell migration and invasion suggesting the significance impact of this protein on cell motility (32). In the present study, we found that treatment of the lung cancer H460 cells with artonin E resulted in the reduction of the cellular CDC42 levels (Figures 5A and B), along with the findings indicating that filopodia protrusions were reduced in the cells treated with artonin E (Figure 4). This substance may, at least in part, inhibit migration and invasion through CDC42 suppression. Even though we did not observe a significant effect of artonin E on the cellular CAV1 level, we and others have previously found the importance of CAV1 in regulating lung cancer cell migration and invasion (16, 33). Recently, CAV1 has been shown to play an important role in cancer metastasis (19). CAV1 was shown to mediate cancer cell migration and invasion in head and neck squamous cell carcinoma (34). Furthermore, overexpression of CAV1 was shown to enhance migration and invasion ability of H460 lung carcinoma cells, while its suppression using ShRNA-CAV1 had the opposite effect (16, 35).
In addition, we have provided evidence indicating that the inhibitory effect of artonin E on cancer cell motility can also be observed in other lung cancer cell models. Three human lung carcinoma cell lines, namely, H292, H23 and A549, were tested with non-toxic concentrations of artonin E and the results indicated that artonin E had similar activities in these cells as those found in H460 cells.
In summary, this investigation reveals that artonin E can inhibit migration and invasion of lung cancer cells via suppression of activated FAK, activated AKT, and CDC42; therefore, artonin E may be a promising agent for anti-metastasis therapy or used as adjuvant with standard therapies to improve survival of patients with cancer.
Acknowledgements
This work is supported by grants from the 90th Anniversary of Chulalongkorn University Fund and the Ratchadaphiseksompot Endowment Fund, Chulalongkorn University. The Authors would like to thank Mr. Krich Rajprasit for proofreading.
- Received May 10, 2013.
- Revision received May 22, 2013.
- Accepted May 28, 2013.
- Copyright© 2013 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.