


Abstract
De-regulated expression of components of the Notch signaling pathway is observed in malignant melanoma. This pathway is activated by catalytic cleavage of the Notch receptor by γ-secretase. Phase-I trials with RO4929097, a potent gamma secretase inhibitor (GSI), and other agents of this class have demonstrated clinical activity in patients with melanoma. An understanding of the mechanisms for de novo sensitivity and resistance to this class of drugs would be critical for future drug development. We treated a panel of Phosphatase and Tensin Homolog (PTEN)-null, -mutant and -wild-type human melanoma cell lines with RO4929097 and evaluated the efficacy alone and in combination with chemotherapy. Although cleaved Notch-1 formation was observed in all the cell lines, RO4929097-induced senescence or apoptosis was achieved only in PTEN-wild-type cell lines in which gamma-secretase inhibition with an induction of PTEN expression and decreased AKT/PKB (protein kinase B) phosphorylation in addition to transcriptional suppression at the Hairy and enhancer of split-1 (HES1) gene promoter. Overexpression of wild-type PTEN in PTEN-null and -mutant cell lines, and studies with isogenic breast cell lines that differ only in PTEN status, confirmed the importance of PTEN expression for conferring tumor cell susceptibility to RO4929097. Furthermore, in PTEN-expressing rapidly accelerated fibrosarcoma 1 (B-RAF)-mutant melanoma cells, RO4929097 enhanced the effect of temozolomide both in vitro and in vivo. These results indicate that tumor cell susceptibility to a GSI, whether alone or in combination with chemotherapy, are reliant upon reducing AKT phosphorylation and hence GSI in combination with chemotherapy may be useful as a new therapeutic approach in treating PTEN-wild-type melanoma.
The incidence of cutaneous melanoma continues to rise within the United States. Despite several recent advances in the treatment of this disease with both rapidly accelerated fibrosarcoma-1 (BRAF) inhibitors and immune modulatory agents that target cytotoxic T-lymphocyte antigen-4 (CTLA-4) and Programmed cell death protein-1 (PD-1), this disease is still considered incurable and new therapeutic approaches are needed. De-regulated expression of Notch signaling pathway components have been observed in many malignancies, including malignant melanoma (1-7). The highly conserved Notch signaling pathway consists of Notch receptors and their ligands, both of which are transmembrane proteins (8). Upon ligand binding, two proteolytic cleavages occur in the Notch receptor, first catalyzed by a disintegrin and metalloproteinase (ADAM) family metalloproteases and the second by γ-secretase. The γ-secretase enzyme complex [presenilin, nicastrin, presenilin enhancer-2 (PEN2) and anterior pharynx-defective-1 (APH1)] (9) releases the Notch-1 intracellular domain (ICD) to translocate to the nucleus, where in association with the DNA-binding protein (CSL) and its coactivator Mastermind-1 (MAM1) (10) gene transcription critical for promoting tumor development and growth is activated. Hence, γ-secretase represents a novel target for cancer therapy. Towards this end, there has been a major effort to develop inhibitors of γ-secretase. Several γ-secretase inhibitors (GSIs) are now in different stages of drug development. The GSI RO4929097 has completed phase-I drug development with clinical activity noted in melanoma patients (11). RO4929097 has been reported to inhibit the growth of melanoma cells both in vitro and in vivo (12). However, the degree of growth inhibition is highly variable and in most cases quite modest. The mechanism(s) of drug resistance and susceptibility remain unknown.
Only few transcriptional targets of Notch-1 have been identified so far and this includes the hairy and enhancer of split-1 (HES) family of transcription factors (13), p21 (14) and Phosphatase and Tensin Homolog (PTEN) (15). AKT/PKB (protein kinase B), a downstream effector of Phosphatidylinositide 3-kinase (PI3K), is persistently activated in many types of human cancer. In melanoma, this can occur through inactivating mutations or deletions of PTEN (16). PTEN is a tumor suppressor gene which encodes a lipid phosphatase that can inhibit the PI3K-AKT signaling pathway by converting phosphatidylinositol triphosphate to phosphatidylinositol diphosphate. The AKT pathway promotes cell growth, cell-cycle progression and cell survival through multiple mechanisms. PTEN exerts its role as a tumor suppressor by negatively-regulating the PI3K-AKT signaling pathway (17).
In T-cell acute leukemia (T-ALL), it has been demonstrated that Notch-1 activates the AKT pathway by down regulating PTEN expression through HES1 (15). Oligonucleotide microarray techniques have demonstrated that loss-of-function mutations or absence of PTEN protein expression in T-ALL is associated with GSI resistance (15). Consistent with the presence of loss-of-function mutations in PTEN, all GSI-resistant T-ALL cell lines analyzed exhibited increased levels of AKT phosphorylation, indicative of constitutive PI3K-AKT activation. These results point to a critical role for the PI3K-AKT signaling pathway in mediating GSI resistance. To help guide future clinical development of RO4929097 for melanoma therapy, we aimed to determine if PTEN impacts upon melanoma cell susceptibility to RO4929097 whether alone or in combination with temozolomide, both in vitro and in vivo.
Materials and Methods
Cell culture. B-RAF mutant cutaneous melanoma cell lines; SK-Mel 32 and SK-Mel 267 (both wild type for PTEN), SK-Mel 39 (PTEN mutant) and SK-Mel 133 (PTEN null) were a generous gift from Alan Houghton, MD (Memorial Sloan Kettering Cancer Center, New York, NY, USA). Cells were maintained in RPMI with 50 U/ml each of penicillin and streptomycin, and 10% heat-inactivated fetal bovine serum, and incubated at 37°C in 5% CO2. MDAMB 468 vector and MDAMB 468 PTEN-inducible cells were a generous gift from Neal Rosen, MD., Ph.D. (Memorial Sloan Kettering Cancer Center, New York, NY, USA) maintained in Dulbecco's Modified Eagle's (DME) media with 50 U/ml each of penicillin and streptomycin, and 10% heat-inactivated fetal bovine serum, and incubated at 37°C in 5% CO2.
Drugs. RO4929097 was provided by Hoffman-La Roche Inc., Nutley, NJ through a cooperative research and development agreement (CRADA) with the National Cancer Institute's (NCI) Cancer Therapy Evaluation Program (CTEP). Temozolomide was purchased from Sigma-Aldrich (St. Louis, MO, USA).
HES1 luciferase assay. The pGL2-Hes-1-luciferase construct was a generous gift from Dr. Raffi Kopan (Washington University, St. Louis, MO, USA). The pGL2 and Renilla vectors were obtained from Promega (Madison, WI, USA). Cell lines were co-transfected with pGL2-Hes-1-luciferase reporter (0.25 μg/well) and the Renilla reporter pRL-CMV in a 10:1 ratio using Fugene-6 transfection reagent (Promega). After 18 h, cells were treated with RO4929097 (1-10 μM) for 48 h and cells were harvested using the Dual-Reporter luciferase assay kit (Stop-and-Glo; Promega), and luciferase activity was quantified on a luminometer (Turner Design, Sunnyvale, CA, USA).
Clonogenic cell proliferation assay. Cells were plated, in triplicate, onto 100-mm dishes at 1,000 per dish and were treated with the indicated concentrations of RO4929097 for 24 h. Cells were then cultured in drug-free medium for 10 to 14 days. The resulting colonies were scored after staining with 0.01% crystal violet (18).
Colorimetric cell proliferation assay. The assay was carried out as per the manufacturer's protocol (Dojindo Molecular Technologies, Inc., Rockville, MD, USA) with minor changes as follows. Briefly, 2,000 cells were plated in 100 μl volume per well of a 96-well plate, and treatments were carried out 24 h after plating. After an incubation period of six days with RO4929097 (5 μM) and temozolomide (300 μM), alone or in combination, the media were replaced with MEM without phenol red with 10% serum and 10% Cell Counting Kit-8 (CCK-8) solution and cells were further incubated at 37°C for 1 to 4 h. In this assay, the amount of formazan dye generated by the activity of dehydrogenases in the cells is quantified and is directly proportional to the number of living cells. The optical density at 450 nm to determine the cell viability was measured using Spectra Max 340 PC (Molecular Devices Corp., Sunnyvale, CA, USA)
Flow cytometry. For flow cytometry (19), the cells were washed and fixed in 75% ice-cold ethanol before staining with propidium iodide (50 μg/ml) containing RNase (5 μg/ml) for the measurement of DNA content. To measure the mitotic population, fixed cells were labeled with monoclonal antibody to phospho Mitotic Protein Monoclonal-2 (MPM2) (Millipore, Billerica, MA, USA), which recognizes specific phosphorylated epitopes present only in mitosis, followed by Alexa Fluor 488 antimouse secondary antibody (Invitrogen, Oregon, USA). Cells were then treated with RNase and propidium iodide. Samples were analyzed on a FACScan (Becton Dickinson, Franklin Lakes, NJ, USA) for cell-cycle distribution and mitotic index (fraction of cells positive for phospho-MPM-2) using Cell Quest software. A total of 10,000 events were examined per sample.
Senescence assay. Cells were plated on 6-well plates, after 48 h of exposure to RO4929097, cells were harvested using senescence assay kit according to the Manufacturer's protocol (Cell Signaling Technology, Inc., Danvers, MA, USA). Briefly, cells were washed with Phosphate Buffered Saline (PBS) and stained for senescence using a β-galactosidase at pH 6 and stored for histology in 70% glycerol. Only senescent cells exhibit β-galactosidase activity at pH 6 assessed by cells stained blue.
si-RNA transfection. Cells were plated on 60-mm plates, and transfections using lipofectamine RNAiMAX (Invitrogen) were performed according to the manufacturer's protocol. The siRNA sequences for Notch-1, nicastrin and control siRNA were purchased from Santa Cruz Biotechnology (Dallas, Texas, USA). Another siRNA for Notch-1 (5’-AAGTGTCTGAGGCCAGCAAGA-3’(20)) was purchased form Dharmacon (Thermo Fisher Scientific, Dharmacon Products, Lafayette, CO, USA). siRNA specific for PTEN was purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Treatments with RO4929097 and temozolomide were performed 18 h after transfection.
Plasmid transfections. For plasmid transfections, cells were plated at 70% confluence, and the transfections were performed on the same day in 60-mm tissue culture plates using Fugene 6 transfection reagent (Roche) in OPTI MEM 1 media (Invitrogen) and 2 μg of pCEP4-PTEN (21) (Ramon Parsons, M.D., Ph.D., Columbia University, New York, NY, USA), or empty-vector, or full-length Notch-1 (a generous gift from Spyros Artavanis-Tsakonas Ph.D., Harvard Medical School, Dept of Cell Biology, Boston, MA, USA) were used per plate. Treatments with RO4929097 and temozolomide were performed 18 h after transfection.
Cell lysate extraction and immunoblotting. Cell lysates were prepared by lysis of both floating and adherent cells in whole-cell lysis buffer [50 mmol/l Tris, pH 7.4, 150 mmol/l NaCl, 1% NP-40, 1 mmol/l EDTA, 0.25% sodium deoxycholate, 0.1 mmol/l Na3VO4, with protease inhibitor cocktail tablet (Roche)], allowed to lyse on ice for 10 min, syringed and cleared by centrifugation in a microcentrifuge at 16,000 ×g for 10 min at 4°C. Fifty micrograms of protein were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto Immobilon membranes (Millipore). After blocking with 5% non-fat milk, membranes were probed with primary antibodies. The following antibodies were used in this study: mouse monoclonal to cleaved poly (ADP-ribose) polymerase (PARP), rabbit polyclonal to cleaved Notch-1, rabbit polyclonal to cleaved caspase 3, rabbit polyclonal to PTEN, rabbit polyclonal to phospho AKT (473), rabbit polyclonal to tubulin and rabbit polyclonal to total AKT, purchased from Cell Signaling. Bound primary antibodies were detected with horseradish peroxidase-conjugated secondary antibodies and visualized by enhanced chemiluminescence reagent (both from GE Healthcare UK Ltd.)
Quantitative fluorescent microscopy (QFM). Cells were collected after drug treatment and fixed in 3% paraformaldehyde. The nuclear morphology of cells was examined using fluorescence microscopy after staining with 4’,6-diamidino-2-phenylindole (DAPI) at a concentration of 25 μg/ml. The number of cells with decondensed, fragmented chromatin was taken as a measure of apoptosis. A minimum of 400 cells were counted for each sample and apoptosis expressed as a percentage as that of untreated cells.
Xenograft studies. Athymic mice bearing SK-Mel 32 tumors (7 mice/cohort, 6-8 weeks old, athymic, female nude mice from Harlan laboratories, USA) of 150 mm3 diameter were either treated with vehicle control, 10 mg/kg of RO4929097 p.o. once daily for 5 days per week for three weeks, 10 mg/kg temozolomide once daily for three weeks, or combination of temozolomide and RO4929097 for three weeks. Tumors were measured every 2 to 3 days with calipers, and tumor volumes were calculated by the formula 4/3 × × r3 [where r=(larger diameter+smaller diameter)/4]. The percentage of tumor regression was calculated as the percentage ratio of the difference between baseline and final tumor volume to the baseline volume. Toxicity was monitored by weight loss. Twenty-four hours after the last treatment on days 8 and 22, and three weeks after the last treatment, one animal from each cohort was sacrificed and the tumors examined for H&E, Ki67 staining, PTEN and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL). Experiments were carried out under an Institutional Animal Care and Use Committee–approved protocol, and institutional guidelines for the proper and humane use of animals were followed (NIH Publication No. 85-23, released 1985).
Histopathology. For immunohistochemical analysis, representative sections of tumors were de-paraffinized, rehydrated in graded alcohols, and subjected to antigen retrieval by microwave oven treatment using standard procedures. H&E staining was carried out using Gill's hematoxylin (Poly Scientific R&D Corp., Bay Shore, NY, USA) for 10 min as per the manufacturer's protocol, followed by counterstaining with eosin (Poly Scientific R&D Corp., Bay Shore, NY, USA) for 4 min. The immunohistochemistry was performed at the Molecular Cytology Core Facility of Memorial Sloan Kettering Cancer Center using Discovery XT processor (Ventana Medical Systems, Oro Valley, AZ, USA). TUNEL staining was performed by proteinase-K treatment, 20 μg/ml followed by TdT-biotin-dUTP labeling mix (Roche) for one hour. The percentage of TUNEL-positive cells from tumor sections was determined by counting at least 100 cells each from at least three randomly selected fields. For Ki-67 (Vector labs, Burlingame, CA, USA) and PTEN (Cell Signaling Technologies Inc., Danvers, MA, USA) staining, after primary antibodies biotinylated goat anti-rabbit IgG Streptavidin-HRP and 3,3’-Diaminobenzidine (DAB) detection kit (Ventana Medical Systems, Oro Valley, AZ, USA) were used according to the manufacturer's instructions.
Results
RO4929097 induces growth suppression of melanoma cells according to PTEN status. The antitumor effect of RO4929097 was evaluated in a panel of cutaneous melanoma cell lines by the clonogenic assay. Figure 1A shows a 40-60% inhibition in clonogenicity for PTEN-expressing SK-Mel 267 and SK-Mel 32 cell lines at concentrations of 1 and 10 μM; PTEN-null SK-Mel 133 and the PTEN-mutant SK-Mel 39 were resistant to the drug. In order to further explore this differential sensitivity according to PTEN status, we examined the relevant downstream signaling pathways. Notch pathway activation leads to the induction of HES1 gene expression; hence, we utilized a HES1-luciferase reporter system to evaluate pathway inhibition elicited by RO4929097. The degree of HES1 promotor inhibition mediated by RO4929097 at 48 h correlated to the relative sensitivity of the cells to the drug, and PTEN status as RO4929097 more effectively inhibited luciferase activity in SK-Mel 32 and SK-MEL 267 (more than 70% inhibition) than in SK-Mel 39 and SK-Mel 133 (50% or less) (Figure 1B).
The effect of RO4929097 on Notch, PTEN and AKT signaling was further confirmed by western blot analysis, as shown in Figure 1C. Despite the difference in the inhibition of luciferase activity and clonogenicity, all the cell lines exhibited almost complete suppression of the cleaved form of Notch-1, suggesting RO4929097 was effective at inhibiting γ-secretase in all the cell lines. However, only in the sensitive cell lines did RO4929097 reduce p-AKT; PARP cleavage was also induced in SK-Mel 32 cells. Although there was no evidence of RO4929097-induced apoptosis in SK-Mel 267, β-galactosidase activity was detected in the RO4929097-treated cells [Figure 1C (ii)], consistent with the induction of senescence. Neither apoptosis nor senescence was detected in the PTEN-null or -mutant cell lines (data not shown).
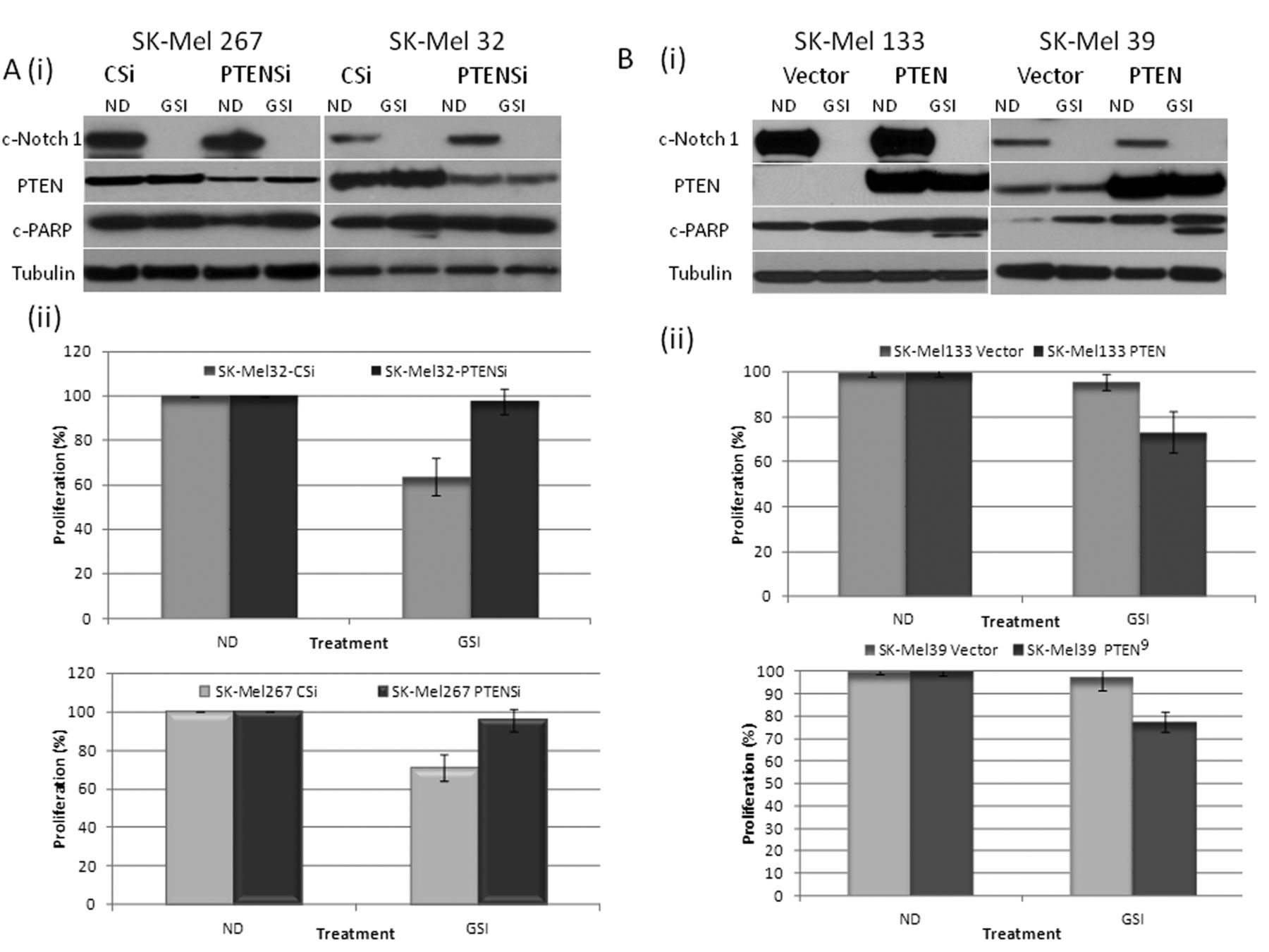
Although no apparent change in PTEN protein was noted after 48 h of GSI exposure of the SK-Mel 32 or SK-Mel 267 cells, at an earlier time point (8 h), there was induction of PTEN and a corresponding decrease in p-AKT (Ser-473) in the setting of partial suppression of cleaved Notch 1 (Figure 1D) which was not observed in SK-Mel 39 and SK-Mel 133 cells. Down-regulating cleaved Notch-1 with specific siRNA in SK-Mel 32 cells (Figure 1E), also resulted in increased PTEN and reduced p-AKT. Similarly, with transient overexpression of full-length Notch-1, an induction of p-AKT was observed. These results suggest that in PTEN-wild-type melanoma cells at baseline, activated Notch-1 induces AKT pathway activation by down-regulating PTEN expression (15). PTEN overexpression sensitizes cells to RO4929097-induced growth suppression. We next explored the hypothesis that PTEN expression mediates tumor susceptibility to RO4929097. Down-regulation of PTEN with specific siRNA in the PTEN-wild-type cell lines SK-Mel 32 and SK-Mel 267 (Figure 2Ai) reversed the antiproliferative effect of the GSI in both cell lines (Figure 2Aii) and blocked PARP cleavage in the GSI-treated SK-Mel 32 cells. Overexpression of PTEN in SK-Mel 133 and 39 cells (null and mutant for PTEN, respectively) increased the antiproliferative effect of RO4929097 in these cell lines and resulted in PARP cleavage (Figure 2B (i) and (ii)).

γ-Secretase inhibitor (GSI) RO4929097 induces growth inhibition by target-specific inhibition. A: Cells were treated with the indicated concentrations of RO4929097. Each data point is derived from triplicates. Data are representative of three independent experiments. Bars, ±SD. B: HES-1 luciferase activity was measured in melanoma cell lines treated with or without 10 μM RO4929097 for 48 h. C: (i) Melanoma cells were treated with 10 μM GSI or 0.1% dimethyl sulfoxide [DMSO, ND (no-drug)] for 24 h and the total protein lysates were probed with antibodies, as indicated. (ii) Staining for senescence-associated β-galactosidase activity in GSI-treated (right) or untreated (left) SK-Mel 267 cells. D: (i) SK-Mel 267 and SK-Mel 32, SK-Mel 39 and SK-Mel 133 cells were treated with 10 μM GSI for 8 h and the total protein lysates were probed with c-Notch-1, c-PARP, c-Casapase-3, PTEN, pAKT and GAPDH antibodies. E: SK-Mel 32 cells were transfected with full length Notch-1 (N1Fl), control siRNA (CSi) or siRNA specific for Notch-1 (N1Si) for 48 h and the total protein lysates were probed with c-Notch-1, PTEN, pAKT and GAPDH antibodies. Tubulin was used to confirm equal loading of protein.
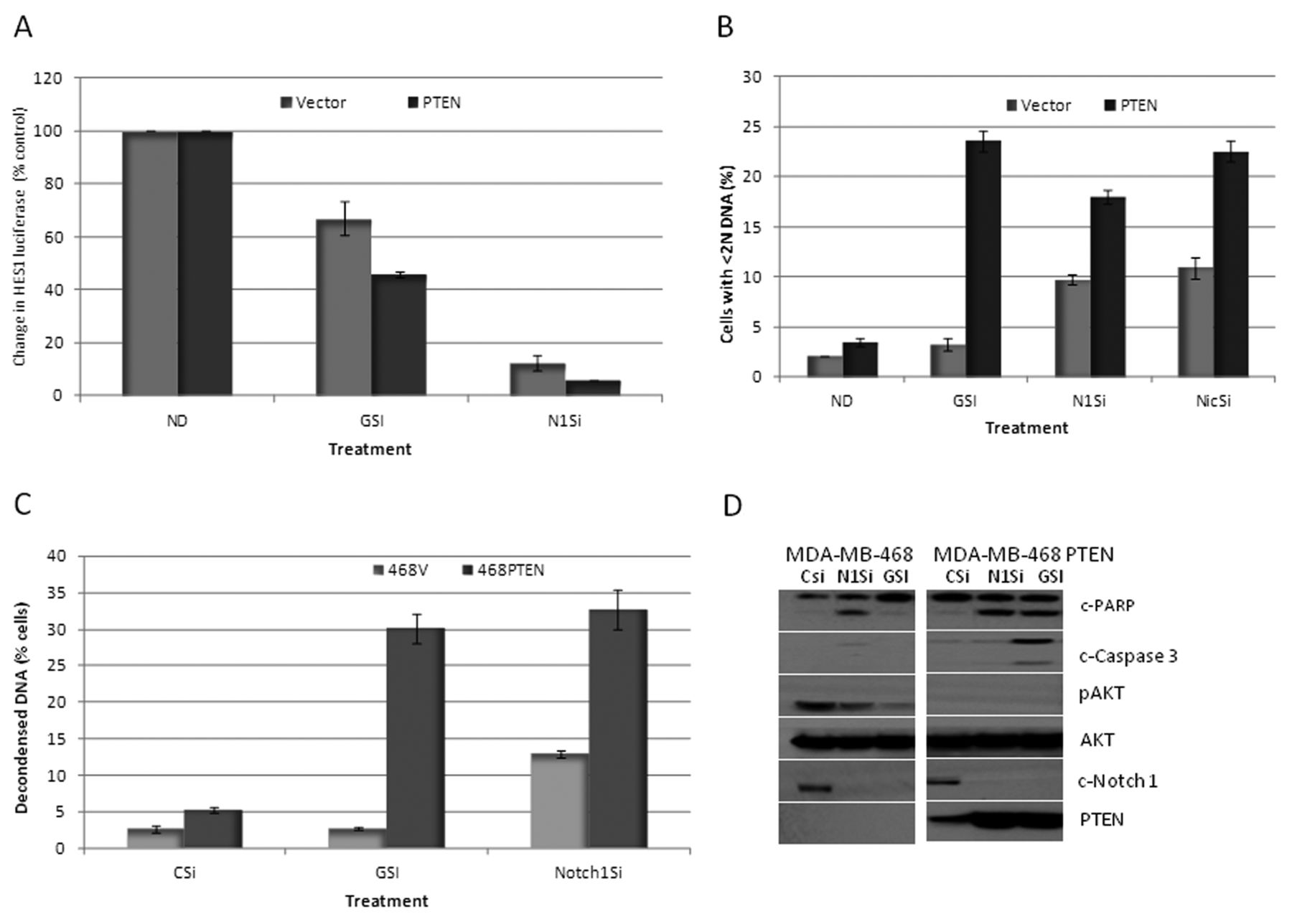
RO4929097 inhibition of Notch-1 is dependent on PTEN expression. In order to confirm the PTEN dependency of the GSI-induced growth suppression and to determine whether this has relevance beyond melanoma, we tested RO4929097 in the MDA-breast cancer cell lines (mutant PTEN) in which either the vector (MDA-468 TR-vector) or wild-type PTEN (MDA-468 TR-PTEN) is controlled by a tetracycline-dependent promoter (22, 23). Cells were treated with the GSI, or Notch-1 was down-regulated by specific siRNA for 48 h. Figure 3A shows that following the use of GSI, the percentage inhibition of HES1 luciferase was greatest in the MDA-MB-468 TR-PTEN cells, as compared to the PTEN-mutant MDA-MB-68 vector-treated controls (60% Vs 40%, p-value=0.01). Both the TR-vector and TR-PTEN MDA-MB-468 cells were sensitive to inhibition of HES1 luciferase activity following down-regulation of Notch-1. However, the inhibition of HES1 luciferase was still relatively greater in the MDA-MB-468-TR-PTEN cells, expressing wild-type PTEN (95% vs. 90% p-value=0.02).
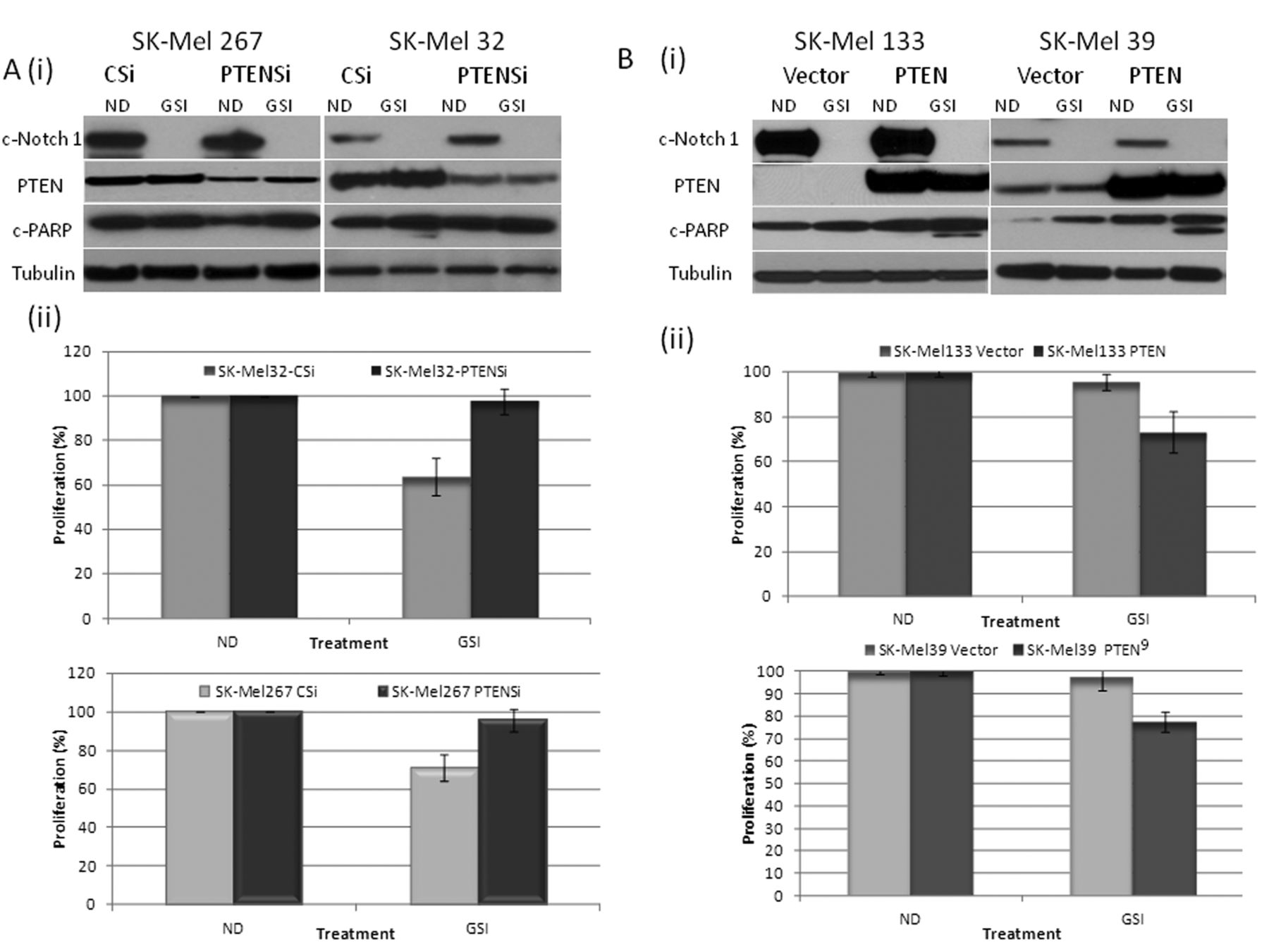
Phosphatase and Tensin Homolog (PTEN) is critical in sensitizing melanoma cells to γ-Secretase inhibitor (GSI) RO4929097. A: (i) SK-Mel 267 and 32 cells were transfected with either control siRNA (CSi) or siRNA specific for PTEN (PTENSi), followed by treatment with or without 5 μM GSI for 48 h and the total protein lysates were probed with c-Notch-1, PTEN, c-PARP and tubulin antibodies. (ii) PTEN down-regulation reverses the effect of growth suppression as determined by the Dojindo Cell Counting Kit, indicating the percentage of growth/proliferation inhibition as compared with control. B: (i) SK-Mel 133 and 39 cells were transfected with either control plasmid or pCEP4-PTEN, followed by treatment with or without 5 μM GSI for 48 h and the total protein lysates were probed with c-Notch-1, PTEN, c-PARP and tubulin antibodies. (ii) Overexpression of PTEN enhances GSI-mediated growth suppression, as determined by the Dojindo Cell Counting Kit, indicating the percentage of growth/proliferation inhibition as compared with control.
This differential effect of the GSI and the siRNA for Notch-1 upon HES1 activity depending on inducible PTEN expression also correlated to differences in the induction of apoptosis. As shown in Figure 3B, FACscan analysis, following treatment with the GSI indicated an increase in the sub-G1 peak from 2% in MDA-MB-468-TR-vector cells to 24% in the PTEN-inducible cells. A similar effect was observed with Notch-1 down-regulation by specific siRNA (9% in the TR-vector cells as compared to 18% in the PTEN-inducible cells). Down-regulation of nicastrin (part of the γ-secretase complex) by specific siRNA treatment also showed a PTEN dependency, with enhanced apoptosis in the TR-PTEN cells (Figure 3B). These effects on apoptosis were confirmed using QFM and by western blot (Figure 3C). By QFM, the GSI and Notch-1 siRNA induced 3% and 12% apoptosis, respectively, of MDA-MB-468-TR-vector control cells vs. 30% and 35% apoptosis, respectively, in the PTEN-inducible cells. Western blot analysis confirmed inhibition of cleaved Notch-1 with both the Notch-1-specific siRNA and the GSI in both cell lines (Figure 3D). However, this was associated with a greater degree of cleaved PARP and cleaved caspase-3 in the PTEN-inducible cells, especially with the GSI. Interestingly, the GSI and the Notch-1 siRNA reduced p-AKT (S473) in the vector control cells, suggesting that pathways also exist independent of PTEN for regulation of p-AKT and induction of apoptosis with inhibition of Notch signaling.
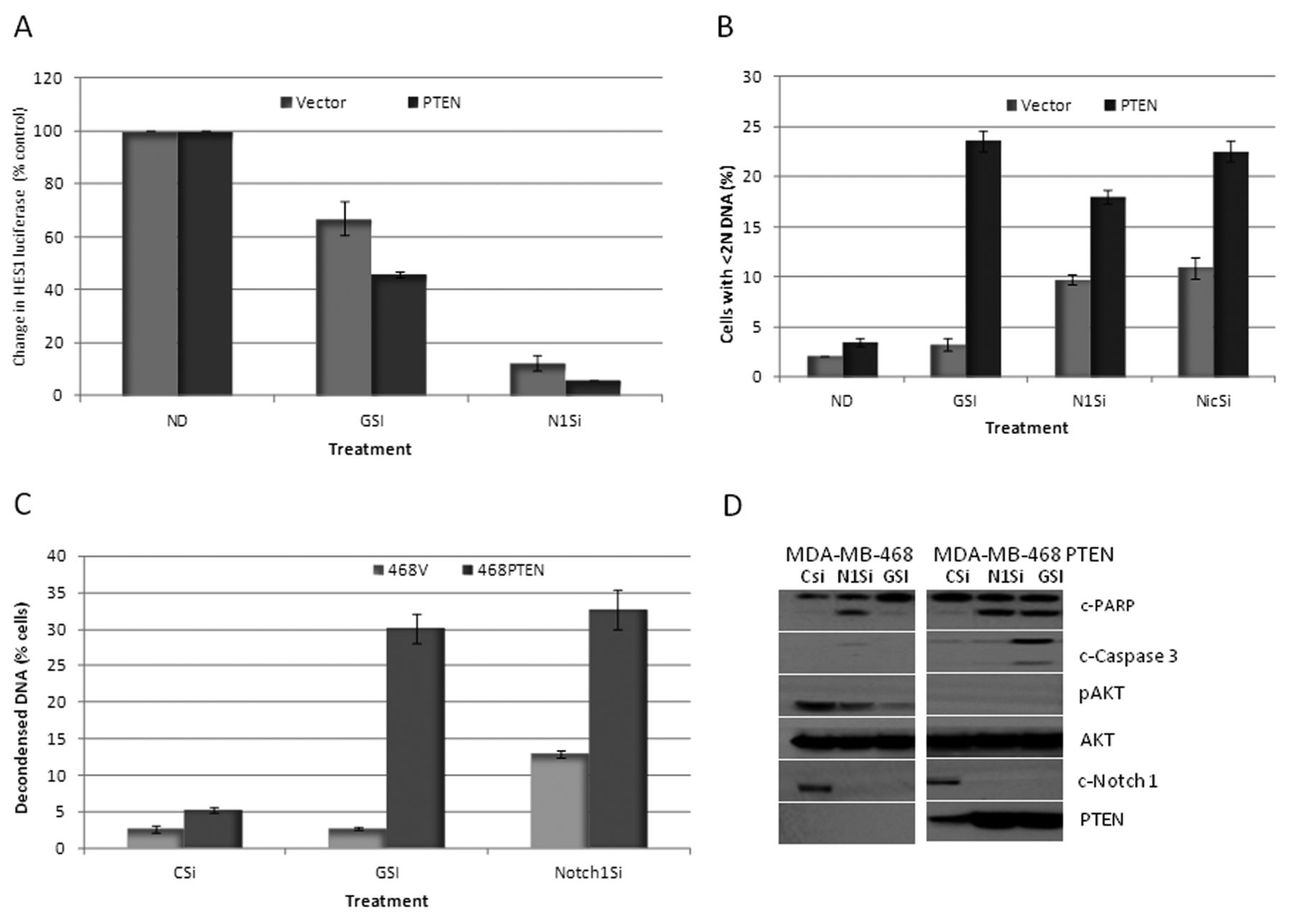
Phosphatase and Tensin Homolog (PTEN) dependency on RO4929097-induced growth suppression. A: HES-1 luciferase activity was measured in MDA-MB-468 vector cells or PTEN-inducible cells treated with or without 10 μM GSI or Notch-1 siRNA for 48 h. B: Percentage of cells with sub-G1 DNA content as determined by FACScan analysis in MDA-MB-468 vector cells or PTEN inducible cells treated with or without 10 μM RO4929097 or Notch-1 siRNA (N1Si) or nicastrin siRNA (NicSi) for 48 h. C: Percentage of apoptosis as determined by quantitative fluorescent microscopy of MDA-MB-468 vector cells or PTEN-inducible cells treated with or without 10 μM GSI or Notch-1 siRNA for 48 h. D: MDA-MB-468 vector cells or PTEN-inducible cells were treated with or without 10 μM GSI or Notch-1 siRNA for 48 h and the total protein lysates were probed with c-Notch-1, PTEN, c-PARP, c-caspase-3, pAKT and total AKT antibodies.
DNA-damaging agent temozolomide enhances the effect of RO4929097 in vitro. We have previously shown that GSIs enhance the effect of chemotherapy, especially of DNA-damaging agents (24). We, therefore, selected to determine whether RO4929097 could enhance the effect of the DNA-damaging agent temozolomide in melanoma cells according to the PTEN status. As shown in Figure 4A, the cells which were wild-type for PTEN (SK-Mel 32 and 267) exhibited enhanced growth inhibition following six days of combination therapy with half the dose (5 μM). On the other hand, cells which were null or mutant for PTEN (SK-Mel 133 and 39) were highly sensitive to single-agent temozolomide, which had little enhanced effect when combined with the GSI. As shown in Figure 4B, the SK-Mel 32 and 267 cells exhibited enhanced PARP cleavage with the combination treatment. This was associated with a decrease in cleaved Notch-1 and inhibition of p-AKT (473), when compared to temozolomide treatment alone. In contrast, in SK-Mel 133 (PTEN-null) and SK-Mel 39 (PTEN-mutant) cells, temozolomide as a single-agent induced PARP cleavage with no further increase with the addition of the GSI (Figure 4B). There was also an increase in p-AKT with temozolomide and the combination therapy, especially in the SK-Mel 133 cells. This effect was further confirmed in the PTEN-inducible MDA-MB-468 cell line. As shown in Figure 4C, in the MDA-MB-468 TR-vector cells, there was no PARP cleavage with single-agent or combination therapy. However, upon induction of PTEN in MDA-MB-468 TR-PTEN cells, there was enhanced apoptosis, as measured by cleaved PARP and an increase in the sub-G1 peak, that was greater than that with temozolomide or the GSI (5 μM as compared 10 μM in single-agent therapy) alone (Figure 4C). Furthermore, this was associated with a further increase in PTEN expression.
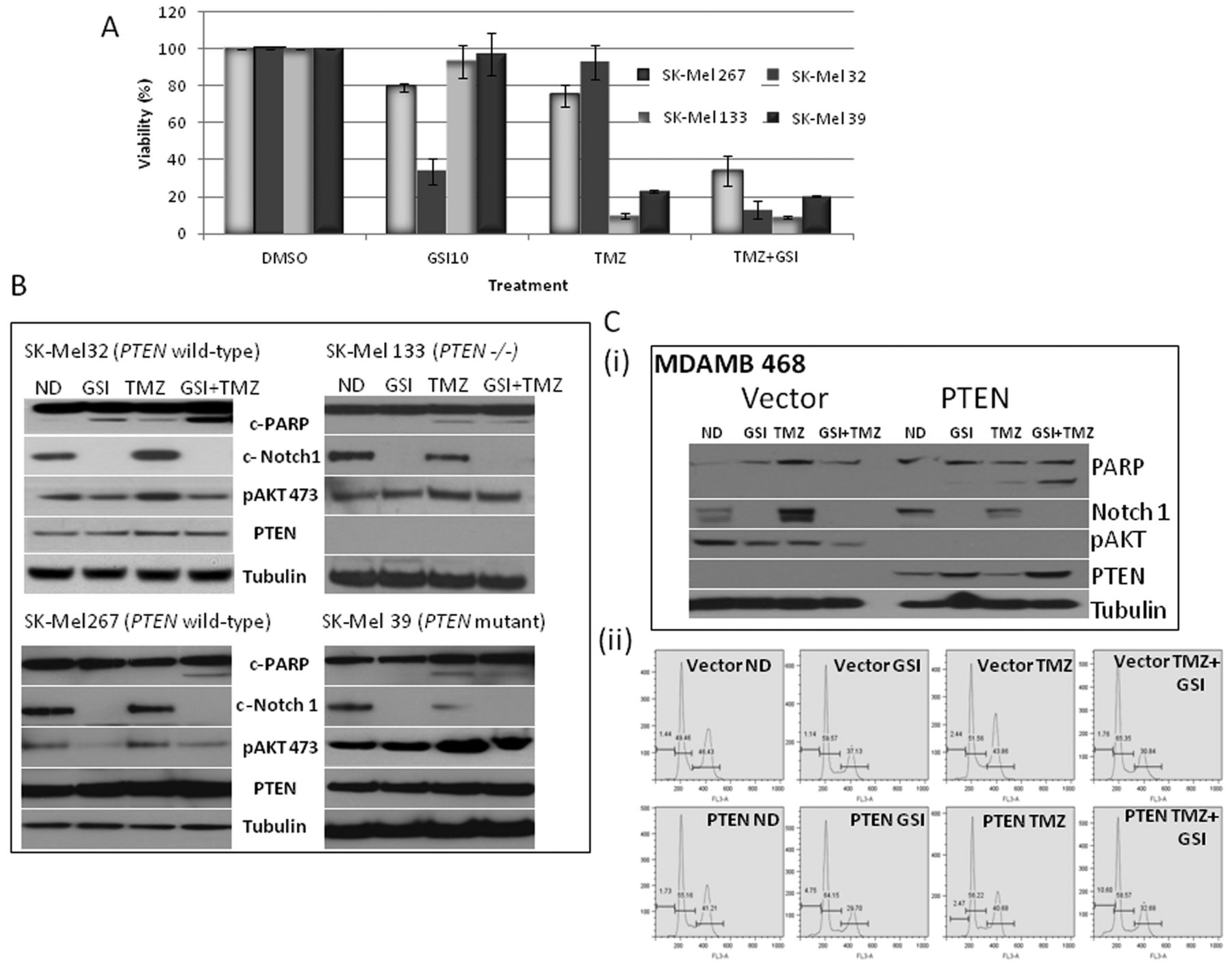
DNA-damaging agent temozolomide (TMZ) enhances the effect of γ-Secretase inhibitor (GSI) RO4929097 in a Phosphatase and Tensin Homolog (PTEN)-dependent manner. A: TMZ enhances the effect of GSI, as determined by measuring cell proliferation using the Dojindo Cell Counting Kit, indicating the percentage of growth/proliferation inhibition as compared with control. B: SK-Mel 32, SK-Mel 267, SK-Mel 133 and SK-Mel 39 cells were treated with DMSO as control (ND), 5 μM GSI-alone, 300 μM TMZ, or their combination and the total protein lysates were probed with c-Notch-1, PTEN, c-PARP, pAKT and tubulin antibodies. C (i): MDA-MB-468 vector cells or PTEN-inducible cells treated with DMSO as control or 5 μM GSI-alone, 300 μM TMZ, or their combination and the total protein lysates were probed with c-Notch-1, PTEN, c-PARP, pAKT and tubulin antibodies. (ii) Fluorescence-activated cell sorting analysis (FACScan) revealed increased sub-G1 peak under the same conditions.
Temozolomide enhances the effect of GSI in vivo. Based on the in vitro data we tested the efficacy of the GSI RO4929097 in combination with temozolomide in PTEN-wild-type SK-Mel 32 xenografts. As shown in Figure 5A, the growth suppression with the combination therapy was greater than that with either single-agent treatment (day 28, p=0.03 (GSI vs. temozolomide+GSI) and p=0.05 (temozolomide vs. temozolomide+GSI). To examine apoptosis, immunohistochemistry was performed for TUNEL and as shown in Figure 5B, apoptosis increased from 2-5% with either vehicle or single-agent therapy to 14% with the combination treatment. Furthermore, as determined by immunohistochemistry and western blot, PTEN protein expression levels also increased with the GSI-alone and in combination with temozolomide (Figure 5C)
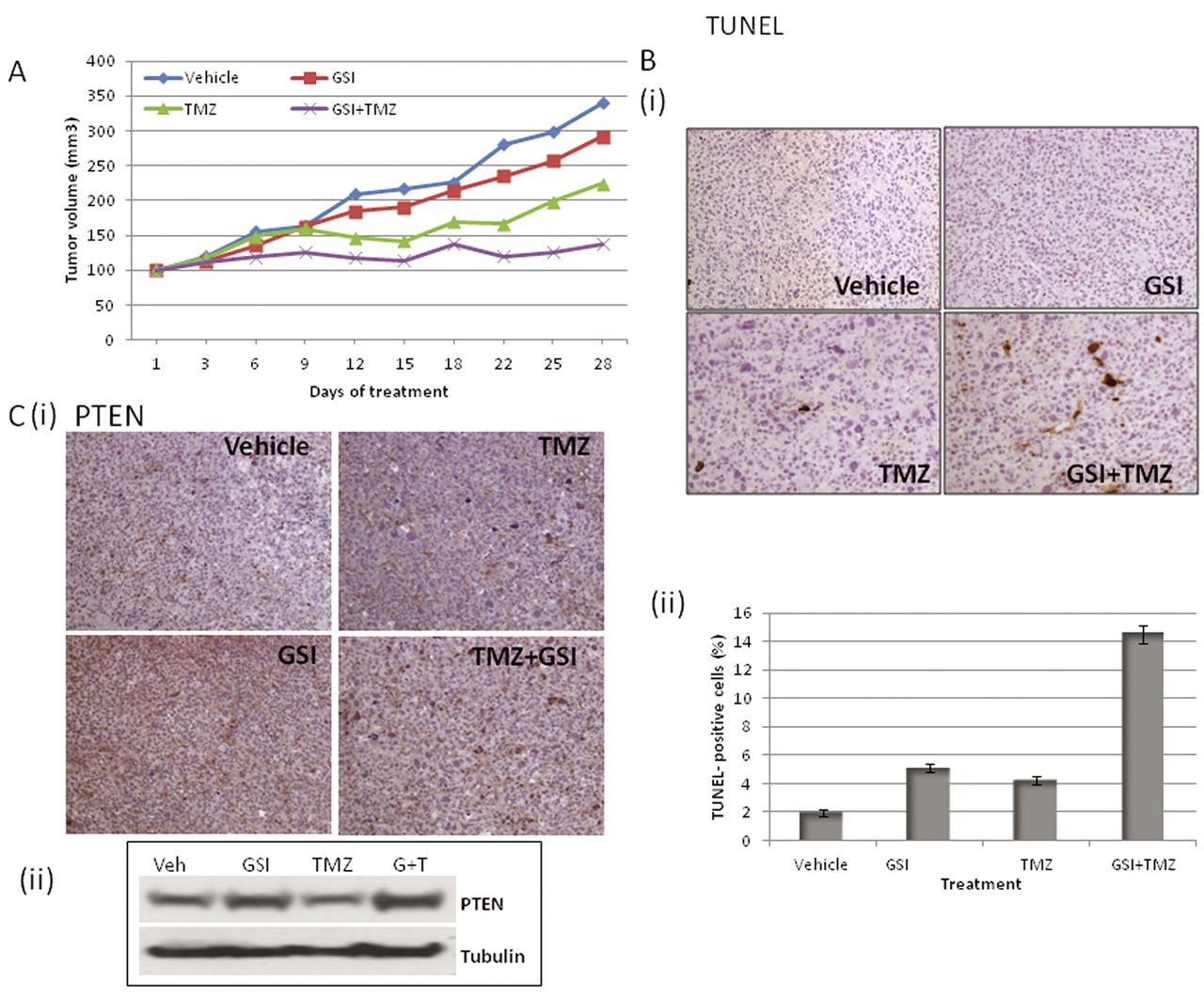
Temozolomide (TMZ) enhances the tumor suppressor effect of γ-Secretase inhibitor (GSI) RO4929097 in vivo. Athymic mice were implanted with SK-Mel 32 tumors and mice (n=7) were treated with GSI (10 mg/kg), TMZ (10 mg/kg), GSI plus TMZ in combination or vehicle, as described in the Materials and Methods. A: Tumor volume was measured every 2 to 3 days and the mean tumor volume was plotted against time in days. B: (i) 24 h after the final treatment, tumors were excised and analyzed by immunohistochemistry for Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) (ii) TUNEL-positive cells were counted from three different fields and the numbers plotted as percentages. C: 24 h after the final treatment, tumors were excised and analyzed by immunohistochemistry for Phosphatase and Tensin Homolog (PTEN) and the xenograft lysates were probed for PTEN protein.
Discussion
Overall, the results of GSI-directed therapy in clinical trials have been disappointing. Even in T-ALL, which is characterized by mutations and activation of Notch signaling, the clinical activity of GSIs has been more modest than expected (25). However, patients with T-ALL also have a high frequency of PTEN mutations. PTEN is transcriptionally-regulated by activated Notch-1. This is believed to be mediated by a transcriptional repressor downstream of Notch which suppresses PTEN expression and activates p-AKT. Our data would suggest that the loss of PTEN could explain the relative resistance of this particular tumor type to GSI therapy. Our results strongly suggest a link between Notch-1 and PTEN expression in melanoma cell lines, and indicate that RO4929097, by specifically inhibiting Notch1-mediated HES1-luciferase activity via blockage of γ-secretase, results in growth suppression in a PTEN-dependent manner. This appears to be mediated, in part, by the induction of PTEN, resulting in inhibition of p-AKT. This results in an increase in apoptosis and senescence and enhancement of the effect of temozolomide both in vitro and in vivo. This has particular clinical relevance, as RO4929097 is now being tested in combination with temozolomide-based chemotherapy in patients with melanoma (clinicaltrials.gov. NCT01196416).
Loss of PTEN expression has been observed in approximately one quarter of V600E-mutant BRAF melanoma cell lines examined (26). In addition, analysis of whole-genome sequence data recently identified phosphatidylinositol-3,4,5-trisphosphate-dependent Rac exchange factor-2 (PREX2), a PTEN-interacting protein and negative regulator of PTEN in breast cancer as a significantly mutated gene with a mutation frequency of approximately 14%. PREX2 somatic mutations generate truncated or variant proteins that gain oncogenic activity in melanoma cells (27). Thus, PTEN loss, or genes that negatively regulate PTEN, appear to play an important role in the biology of melanoma.
In the phase-I trial of R0929097, a patient with widespread metastatic melanoma had a minor response by RECIST criteria and prolonged stable disease was seen in patients with several other tumor types. This included a patient with colorectal cancer that had neuroendocrine features (11). There were also suggestions of activity in epitheloid sarcoma. However, the PTEN status of these patients was not reported. In this clinical trial there was an attempt to identify a putative biomarker of response. With R04929097 a weak-to-moderate association between drug level and changes in several putative markers of Notch inhibition were reported. These included changes in Aβ-40 protein in plasma, Vascular Endothelial Growth Factor Receptor-2 (VEGFR-2) protein in plasma, and HES-1 mRNA expression in hair follicles, as well as changes in notch signaling from limited tumor sampling. However, none of these biomarkers were found to be predictive of response to therapy. Plasma markers of Notch inhibition, in general, have been difficult to measure in a quantifiable and reproducible way. In addition, in the phase-I clinical trial of the GSI MK-0752, the investigators reported a correlation between a 9-gene Notch signature score and pharmacological exposure to MK-0752 in individual patients (28). This pharmacodynamic biomarker of Notch inhibition was first derived from a healthy volunteer study and included changes in Notch transcriptional expression in pre- and post-treatment hair follicles at 8 h (30). Whether this in any way correlated to target inhibition in the tumor remains unknown.
With limited clinical activity and no validated biomarker at hand, there remain concerns that GSIs may be abandoned before a successful biomarker can be established. However, with proper patient selection it may be possible to select a patient population (PTEN-wild-type) that is sensitive to RO4929097 or other GSI-targeted-therapy. With the emergence of drug resistance to BRAF-targeted therapies, and with essentially no effective therapy for patients with BRAF-wild-type, the identification of a patient population which is sensitive to a GSI-targeted therapy, especially as part of combination therapy with temozolomide, could have significant implications in the development of this class of drugs in this field.
Footnotes
-
This article is freely accessible online.
- Received February 25, 2013.
- Revision received March 13, 2013.
- Accepted March 14, 2013.
- Copyright© 2013 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.