


Abstract
Background: Cancer stem cells (CSCs) are resistant to radiotherapy and are responsible for tumor recurrence of various malignant tumors, including prostate cancer. Materials and Methods: In order to define the radioresistance mechanism of prostate CSCs, their proliferative activity, cell cycle distribution, expression of CD133 stem cell marker, reactive oxygen species (ROS) production, and DNA repair efficiency were examined using prostatospheres and adherent LNCaP cells as a model of prostate CSC and bulk model of differentiated cells, respectively. Results: Compared to adherent cells, prostatospheres exhibited greater number of low-to-intermediate ROS-producing cells and CD133-positive cells. Prostatospheres showed higher expression of DNA repair proteins after ionizing radiation (IR). Conclusion: Low vulnerability to ROS-induced cellular damage and the efficient repair of IR-induced DNA injury may explain the radioresistance of prostate CSCs. Therefore, increasing ROS-induced cytotoxicity and inhibition of DNA repair in prostate CSCs may help achieve complete eradication of prostate CSCs by radiotherapy.
- Prostatic neoplasms
- neoplastic stem cells
- radiotherapy
- reactive oxygen species
- DNA repair
- radioresistance
Radiotherapy is one of the first-line treatments for clinically-localized prostate cancer-but approximately 20-50% of patients with prostate cancer experience tumor recurrence after radiotherapy (1). Recently, cancer stem cells (CSCs) have become the center of growing attention, partly because they are resistant to conventional anticancer treatments, including radiotherapy, and considered as a cause of tumor recurrence (2-4).
According to the CSC model, a tumor is a heterogeneous cell population composed of a small population of CSCs and a bulk of differentiated cells. CSCs can initiate tumors via their self-renewal capacity and multipotency (5-8). As a result, even a few residual CSCs surviving conventional anticancer therapy can cause tumor recurrence (2-4). CSCs have been isolated by flow cytometric sorting using various cell surface markers, including cluster of differentiation 133 (CD133) and CD44. The CSC population can also be enriched by growing cells in non-adherent defined media as cell balls, such as prostatospheres of prostate cancer cells (7-9).
Ionizing radiation (IR) kills tumor cells by damaging DNA bases and cleaving the DNA backbone, which results in DNA double-strand breaks (DSBs) (10). IR also generates reactive oxygen species (ROS), which are formed from oxygen and are the best-known radiosensitizer enhancing radiation-induced cellular damage (11). In spite of radiation-induced injury, some cells survive, resulting in tumor recurrence. This is explained in part by the repair of the IR-induced DNA injuries and can be affected by various factors (10). DSBs are known as the most lethal among IR-induced DNA injuries. However, DSBs can be repaired by the non-homologous end-joining (NHEJ) mechanism, which can be measured by the expression of phosphorylated histone H2AX on serine 139 (γ-H2AX) (10). After IR, the Ku heterodimer (Ku70/80) detects DSBs and acts a central element in NHEJ by interacting with and by recruiting repair proteins to sites of DNA damage. The cell-cycle phase of cancer cells also influences their sensitivity to radiation. Tumor cells are radioresistant when they are in the G0 and S phases, whereas they are radiosensitive in the G2 and M phases (12).
According to our recent report, prostate CSCs are initially damaged by IR but recurrent prostate cancer following IR can lead to increased numbers of CSCs (13); this study suggests that prostate CSCs are radioresistant but the mechanism of this radioresistance has not yet been defined.
Materials and Methods
Cell culture and preparation of prostatospheres. The prostate cancer cell line LNCaP was purchased from the American Type Culture Collection (Manassas, VA, USA) and maintained in RPMI-1640 supplemented with 10% fetal bovine serum, 2 mM L-glutamine, penicillin and streptomycin in a humidified environment at 37°C and 5% CO2. Prostatospheres were generated by plating cells at a low density (1,000 to 5,000 cells/ml) in serum-free medium, which consisted of Dulbecco's Modified Eagle's Medium: Nutrient Mixture F-12 (DMEM:F12) plus 10 ng/ml basic fibroblast growth factor (Invitrogen, Grand Island, NY, USA), 20 ng/ml epidermal growth factor (Prospec, Rehovot, Israel), 5 mg/ml insulin, 0.4% bovine serum albumin (Sigma, Steinhein, Germany) and B-27 supplement (Gibco, Grand Island, NY, USA). Prostatospheres were cultured for six days prior to use. A single-cell preparation from prostatospheres was produced by enzymatic dissociation using trypsin-EDTA (Gibco) and subsequent neutralization using serum-free trypsin-neutralizing solution.
Radiation treatment. Cell cultures were irradiated with 10 Gy at a dose rate of 2 Gy/min at room temperature with a 6-MV photon beam generated by a linear accelerator (CLINAC iX®; Varian Medical Systems, Palo Alto, CA, USA). Corresponding controls were sham-irradiated.
Western blotting. Total protein was isolated from cultured cells using RIPA buffer (Thermo Scientific, Rockford, IL, USA) and protein concentration was measured using a Bio-Rad Protein Assay Kit (Bio-Rad Laboratories, Hercules, CA, USA). After electrophoresis in 4%-20% tris-glycine gel (Bio-Rad Laboratories), proteins were transferred to a polyvinylidene fluoride membrane, blocked in 5% nonfat dry milk in tris-buffered saline (TBS) with 0.1% Tween 20 (TBST) for one hour, and then incubated with the primary antibodies listed below at 4°C overnight. After washing with TBST three times, the membranes were treated with horseradish peroxidase-conjugated secondary antibodies for one hour. After washing, the membranes were developed with the West Pico Chemiluminescent Substrate (Pierce, Rockford, IL, USA) and exposed to Medical X-ray Film Blue (AGFA, Mortsel, Belgium). Primary antibodies used for this study were as follows: phosphohistone H2AX (Ser 139) (Millipore, Temecula, CA, USA), Ku70 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), Ku80 (also known as Ku86; Santa Cruz Biotechnology), and β-actin (Sigma, Saint Louis, MO, USA).
Flow cytometric analyses for intracellular ROS, cell cycle distribution, and CD133 expression. Cells were trypsinized, fixed with 70% ethanol at 4°C overnight and then incubated with 40 μg/ml propidium iodide and 100 μg/ml RNAse A at room temperature for 30 min. The number of cells in each cell-cycle phase was evaluated with a fluorescence-activated cell sorter (FACS) Calibur System (Becton Dickinson, San Jose, CA, USA). For the analysis of intracellular ROS, cells were loaded with 10 μM 2’7’-dichlorofluorescein diacetate (DCF-DA) (Invitrogen, Carlsbad, CA, USA), incubated at 37°C for 30 min, and then immediately analyzed by flow cytometry. In the case of CD133 and ROS double-staining, cells were first treated with antibodies to CD133 [CD133/1 (AC133); Miltenyi Biotec, Auburn, CA, USA] for 15 minutes at room temperature and then treated for ROS as described above.
Cell block preparation and immunohistochemistry. Adherent cells and prostatospheres of LNCaP cells were harvested and fixed in 70% alcohol. After paraffin embedding, the cell blocks were sectioned and stained with hematoxylin and eosin (H&E). For immunohistochemical staining, the sections were stained with an antibody to Ki-67 (Thermo Scientific) as previously described (14).
Statistical analysis. Results are expressed as the mean±standard deviation (SD) of at least triplicate determinations. Statistical comparisons were based on a two-tailed Student's t-test. Differences were regarded as statistically significant at p-values <0.05.
Results
No differences in proliferative activity and cell-cycle distribution was found between prostatospheres and adherent cells. Adherent cells and prostatospheres were examined for the expression of a widely used proliferation marker, Ki-67, which is expressed in the nuclei of cycling cells (G1, S, G2, and mitosis), but is not expressed in resting cells (G0).
Most adherent cells and prostatospheres expressed Ki-67, indicating the high proliferative activity of both cell types (92.0% and 93.7%, respectively). There was no statistical difference between them with respect to proliferation (p=0.58) (Figure 1A-E).
Likewise, there was no statistical difference in cell-cycle distribution between adherent cells and prostatospheres in G0/G1 phase (49.7% versus 52.7%, p=0.73), S phase (44.1% versus 37.1%, p=0.53) or G2/M phase (6.2% versus 10.3%, p=0.21) (Figure 1F).
Low levels of intracellular ROS in prostatospheres. Regardless of whether the cells were adherent or in prostatospheres, three types of cells with different levels of intracellular ROS were evident as follows: those with low, intermediate, and high ROS production (Figure 2A and B). In comparison to adherent cells, prostatospheres demonstrated fewer cells with high ROS production and more with intermediate/low ROS levels and a higher proportion of CD133-positive cells (0.26% versus 0.07%) (Figure 2A and B).
High expression of DNA repair proteins in prostatospheres. To compare the propensity for DNA repair in prostate CSCs with that in differentiated cells, the expression of DNA repair proteins γ-H2AX, Ku70, and Ku80 was examined in prostatospheres and adherent cells at various time points before and after IR with a single-dose of 10 Gy.
As expected, IR induced DNA repair activity in both adherent cells and prostatospheres, as evidenced by the increase of γ-H2AX expression after the radiation (Figure 3). Interestingly, the γ-H2AX expression in prostatospheres was higher than that of adherent cells. Furthermore, γ-H2AX expression was even increased in prostatospheres at two hours after IR while it was decreased in adherent cells at that time. Expressions of Ku70 and Ku80 proteins followed similar trends to that of γ-H2AX.

Morphology, proliferative activity and cell-cycle distribution of adherent cells and prostatospheres. Hematoxylin-eosin staining and Ki-67 immunohistochemical staining of cell block preparations from adherent (A and C) and prostatospheres (B and D). The proportions of Ki-67-positive cells (E) and cell-cycle distribution (F) are summarized. Bars represent the mean±SD. Data shown are representative of at least three independent experiments. Original magnification, ×1000 for A and B, ×400 for C and D.
Discussion
According to a previous report, prostate CSCs are damaged and decreased in number immediately after IR, but their numbers increase in recurrent tumors after long-term recovery (13). The previous study suggests that prostate CSCs possess a greater capacity for recovery after IR-induced cellular damage (13). In this study, we have demonstrated that the radioresistance mechanism of prostate CSCs includes low vulnerability to ROS-induced cellular damage in addition to a high repair capacity for IR-induced DNA injury.
ROS are chemically reactive, oxygen-containing molecules and include superoxide (O2−), hydrogen peroxide (H2O2) and the hydroxyl free radical (HO•). They are generated as a natural by-product of normal oxygen metabolism. In spite of being short-lived, ROS interact rapidly with various intracellular biomolecules such as DNA (15). ROS generated near DNA (within 2 nm) cause more DNA damage than the direct ionization of DNA strands (16). Therefore, normal cells regulate intracellular ROS content within a non-toxic range by balancing ROS-generating and -scavenging systems (17).
Maintaining ROS at a low level is also critical for stem cell function (18). The ROS defense system is operated via the adhesion molecule CD44, which is also a commonly used stem cell marker and is highly expressed in CSCs (19). CD44v, a variant isoform of CD44, increases antioxidative capacity by promoting the synthesis of intracellular glutathione, which functions as an antioxidant, reducing intracellular ROS levels (19). In fact, CSCs of leukemia and breast cancer have a low level of intracellular ROS and express the CSC marker CD44 (18, 20). CD44 is also a surface marker of prostate CSCs and is highly expressed in these cells (8). Therefore, the high expression of CD44 in prostate CSCs at least partly explains the low level of intracellular ROS in prostate CSCs and their low vulnerability to ROS-induced cellular damage.
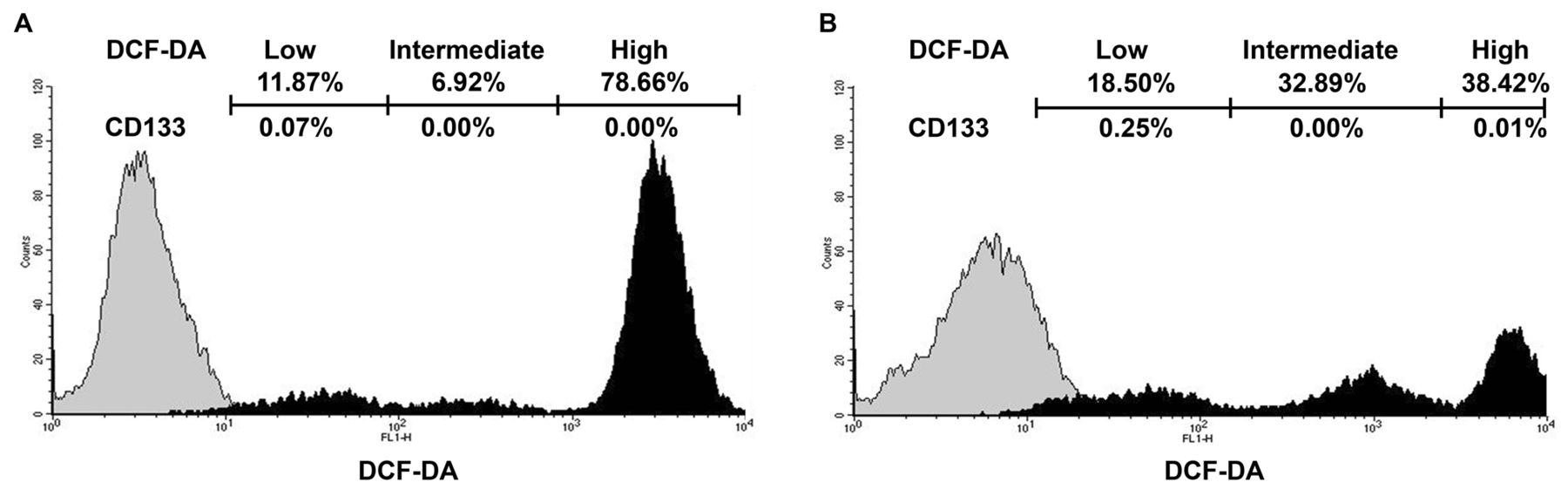
Analysis of intracellular reactive oxygen species (ROS) activity in adherent cells and prostatospheres. The intracellular ROS activity was measured before (grey) and after (black) 2’7’-dichlorofluorescein diacetate (DCF-DA) staining of adherent cells (A) and prostatospheres (B) of LNCaP cells. Three groups with different amounts of DCF-DA-positive cells were present: Low, intermediate and high. The percentage of DCF-DA-positive cells or CD133-positive cells in the three groups is indicated in the upper portion of Figure 2. Data shown are representative of at least three independent experiments.
Prostate CSCs appear to have a high capacity for repair of IR-induced DNA injury as evidenced by the high expression of DNA repair proteins γ-H2AX, Ku70, and Ku80 in prostatospheres after IR. Previous reports have shown that CSCs in various types of tumors possess efficient DNA repair systems (3, 21). In brain tumors, CSCs repair IR-induced DNA damage more effectively than differentiated tumor cells through the activation of DNA checkpoint proteins (3). The efficient DNA repair mechanisms of CSCs can be attributed to B-lymphoma Mo-MLV insertion region 1 homolog (BMI1). As a polycomb protein, BMI1 is essential for self-renewal cell division of stem cells and is enriched in CSCs of various tumor types, including prostate cancer (8, 22-25). BMI1 is rapidly recruited to DNA damage sites and co-purifies with DSB repair proteins (23, 24). Loss of BMI1 leads to the impaired repair of DNA DSBs, whereas BMI1 overexpression enhances the recruitment of repair proteins to chromatin and increases resistance to radiation (23, 24). Therefore, the high expression of BMI1 in prostate CSCs may explain the efficient repair of IR-induced DNA injury in these cells.
Conventional chemotherapy and radiotherapy target rapidly proliferating cells. The quiescent slow-cycling phenotype is a feature of CSCs and explains their resistance to conventional anticancer treatment (26). In the present study, however, most adherent cells and prostatosphere cells were cycling rapidly, as evidenced by their high Ki-67 expression. In contrast, Ki-67-positive tumor cells usually make up less than 10% of the total cell population in human prostate cancer tissues (27, 28). This discrepancy could be due to differences between the in vivo condition of prostate cancer in the human body and the in vitro experimental conditions of the LNCaP culture. Cell lines such as LNCaP are established from a selected subpopulation of rapidly growing tumor cells, in which slowly-growing differentiated cells lose out (29). In fact, the high proliferative activity of LNCaP cells, as evidenced by a Ki-67 positivity of more than 90%, has been previously documented (30). Therefore, such a difference is one of the limitations of this study and should be resolved by examining prostate CSCs and bulk cells of primary human prostate cancer. Furthermore, the low vulnerability of prostate CSCs to ROS-induced cellular damage and their efficient repair of IR-induced DNA injury should also be confirmed in primary human prostate cancer cells.

Expression of DNA repair proteins in adherent cells and prostatospheres before and after ionizing radiation. The expression of DNA repair proteins, phosphorylated histone H2AX (γ-H2AX) and Ku heterodimer (Ku70 and Ku80), was evaluated by western blotting. Adherent cells and prostatospheres of LNCaP cells were irradiated with a single dose (10 Gy) of ionizing radiation and were then harvested at the indicated time points. DNA repair protein expression was higher in prostatospheres than in adherent cells at each time point after ionizing radiation. β-Actin was used as a loading control.
Taken together with the results of our previous study, the present study indicates that the increased survival of prostate CSCs after IR can be attributed to their low vulnerability to ROS-induced cellular damage and their efficient repair of IR-induced DNA damage. Thus, in order to achieve the complete eradication of prostate CSCs by radiotherapy, a novel strategy that increases ROS-induced cellular damage or inhibits DNA repair in these cells is required.
Acknowledgements
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (2010-0013063).
Footnotes
-
Conflicts of Interest
None.
- Received August 2, 2013.
- Revision received September 14, 2013.
- Accepted September 16, 2013.
- Copyright© 2013 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- The soluble guanylyl cyclase pathway is inhibited to evade androgen deprivation-induced senescence and enable progression to castration resistance
- Predictors of Overall and Disease-Free Survival in Metastatic Castration-Resistant Prostate Cancer Patients Receiving 225Ac-PSMA-617 Radioligand Therapy
- Stem cells and the impact of ROS signaling