


Abstract
Background: Pharmacological inhibition of the phosphoinositide 3-kinase (PI3K)/Akt pathway prevents G1 cell cycle progression into S, resulting in G1 accumulation. The hypothesis that this arrest might negatively impact on chemotherapeutic agents primarily effective in S, G2 or M-phase was investigated. Materials and Methods: Inhibition of PI3K/Akt pathway signaling via LY294002 and Akti-1/2 was demonstrated by immunoblotting. Cell cycle progression was determined by flow cytometric analysis. Cell proliferation was assayed using the XTT cell viability assay. The Chou and Talalay median effect principal was used to evaluate drug interaction. Results: In SKOV3 and IGROV1 human ovarian cancer cells, LY294002 and Akti-1/2 increased the percentage of cells in G1 and reversed the cell cycle effects of cisplatin, paclitaxel, gemcitabine and topotecan. Pathway blockade synergistically enhanced the cytotoxicity of cisplatin and paclitaxel, but antagonized gemcitabine and topotecan effects. Conclusion: Pharmacological PI3K/Akt inhibition antagonizes the efficacy of chemotherapeutic agents primarily effective in the S or G2-phase of the cell cycle.
CI, combination index; CI75, CI at ED75; ED, effective dose; NS, not significant.
Ovarian cancer affects 22,000 women a year and is the 5th leading cause of female related deaths in the US (1). The current therapy for ovarian cancer consists of surgical removal of the tumor followed by chemotherapy (2-4). First line chemotherapy is comprised of a platinum-taxane combination, while second line treatment may include agents such as gemcitabine or topotecan.
Activation of the oncogenic phosphoinositide 3-kinase (PI3K)/Akt pathway has been demonstrated in ovarian cancer cell lines and ovarian tumor tissue (5-14). In this signaling cascade activated PI3K phosphorylates phosphoinositide 3,4 bisphosphate (PIP2) to phosphoinositide 3,4,5 trisphosphate (PIP3), which acts to recruit the serine/threonine kinase Akt to the membrane. Phosphorylated Akt activates several downstream targets affecting various biological processes, including cell proliferation and survival (15-17). The PI3K/Akt pathway is antagonized by the tumor suppressor protein phosphatase and tensin homolog (PTEN) via its dephosphorylation of PIP3. Recent molecular data including mRNA expression, microRNA expression, promoter methylation and DNA copy number in 489 high-grade serous ovarian adenocarcinomas and the DNA sequences of exons from coding genes in 316 of these tumors from The Cancer Genome Atlas project have been analyzed (18). Amplification of PIK3CA, the gene encoding the p110α subunit of PI3K, Akt1 and Akt2 was found in 18%, 3% and 6% of the samples, respectively, while Pten deletions were detected in 7%.
Inhibition of the PI3K/Akt pathway has been proposed as a strategy to sensitize tumors to chemotherapy (19, 20). The PI3K/Akt pathway regulates the G1/S cell cycle transition via transcriptional regulation of cell cycle proteins and reduction of cell cycle inhibitors (21, 22). Pharmacological inhibition of the pathway prevents G1 progression into S, resulting in an accumulation of cells in G1. This cell cycle arrest might impact on the cytotoxic effects of chemotherapeutic agents that are primarily effective in the S, G2 or M-phases of the cell cycle. To investigate this hypothesis four chemotherapeutic agents with different mechanisms of action that are commonly used for the treatment of ovarian cancer, namely cisplatin, paclitaxel, gemcitabine and topotecan were chosen (23). Cisplatin is a DNA alkylating agent that causes DNA breaks, paclitaxel inhibits depolymerisation of microtubules during cell division, gemcitabine is a nucleoside analog that causes inhibition of DNA synthesis once incorporated into the DNA, and topotecan is a topoisomerase I inhibitor that prevents religation of nicked DNA during DNA synthesis. Cisplatin is rather cell cycle unspecific and paclitaxel is most active in the M-phase, while both gemcitabine and topotecan elicit their effects mainly during S-phase.
The effect of PI3K/Akt pathway inhibition in combination with cisplatin, paclitaxel, gemcitabine or topotecan, on cell cycle progression and cell proliferation was investigated in SKOV3 and IGROV1 human ovarian cancer cells that both have activating PI3K/Akt pathway mutations. SKOV3 cells contain an activating mutation (H1047R) in the PIK3CA gene. IGROV1 cells harbor a heterozygous deletion mutation in the Pten gene that results in low expression levels of the PTEN protein.
Materials and Methods
Cell culture. The SKOV3 (American Type Culture Collection, Manassas, VA, USA) and IGROV1 (National Cancer Institute, Frederick, MD, USA) cell lines were maintained in Dulbecco's Modified Eagle's Medium (DMEM) (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS). The cells were grown at 37°C in a humidified atmosphere containing 5% CO2 and 95% air.
Immunoblot assay. The cells were grown for 3-4 days on 10 cm dishes, serum-starved overnight, then treated with or without the PI3K inhibitor LY294002 or an allosteric inhibitor of Akt1 and Akt2 phosphorylation, Akti-1/2 (24-26) (both from Sigma Aldrich; St. Louis, MO, USA) for 24 h in the presence of serum. The cells were then lysed with 1× cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA) and the protein concentration of the clarified cell lysates was determined and normalized using the bicinchoninic acid (BCA) protein assay reagent (Pierce Biotechnology, Rockford, IL, USA). The lysates were mixed with 4× Laemmli buffer (200 mM Tris–HCl pH 6.8, 1 mM EDTA, 6% SDS, 2 mM EDTA, 4% 2-mercaptoethanol and 10% glycerol) and analyzed by SDS-PAGE followed by immunoblotting. Primary antibodies were obtained from Cell Signaling Technology (phospho-4E-BP1 (eukaryotic translation initiation factor 4E-binding protein 1) (T37/46), phospho-Akt (S473), Akt, phospho-S6 (S240/244) and S6). Immuno-reactive bands were visualized by chemifluorescence (ECL-Plus; GE Healthcare Biosciences, Piscataway, NJ, USA) detection of horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit secondary antibodies (both from Cell Signaling Technology) and captured with a Typhoon® 9400 scanner (GE Healthcare Biosciences). The blots were stripped and reprobed for β-actin (Santa Cruz, CA, USA) as a loading control.
Cell viability assay. Cell proliferation was assayed using the XTT (2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide inner salt) cell viability assay. The cells were seeded in phenol red-free growth medium in 96-well microtiter plates at 5×103 cells/well and incubated overnight at 37°C. Cisplatin (Sigma Aldrich), paclitaxel (Fisher Scientific, Houston, TX, USA), topotecan (Sigma Aldrich) or gemcitabine (Sigma Aldrich), with or without LY294002 or Akti-1/2, was added the next day and the cells were incubated for 72 h at 37°C. XTT (1 mg/ml) (Invitrogen) and PMS (phenazine methosulphate; 1 mg/ml) (Sigma Aldrich) were added, and the metabolism of XTT was measured over time at 450 nm on an absorbance microplate reader (ELx800, Bio-Tek Instruments, Winooski, VT, USA).
Cell cycle flow cytometric analysis. Cell cycle progression was studied using flow cytometry. The cells were plated at 5×105 cells/well in 6-well microtiter plates and incubated overnight at 37°C. The following day fresh medium was added with or without treatment (cisplatin, paclitaxel, topotecan or gemcitabine, with or without LY294002 or Akti-1/2) for 24 h. Chromosomal DNA was stained using a propidium iodide (100 μg/ml) (Invitrogen), RNAse A (0.02 mg/ml) (Fisher Scientific) and Triton X-100 (0.3%) solution, and analyzed on a FACScan flow cytometer (BD Biosciences, San Jose, CA, USA) using the CellQuest software package (BD Biosciences). Flow cytometry was performed at the UCLA Jonsson Comprehensive Cancer Center and Center for AIDS Research Flow Cytometry Core Facility that is supported by the National Institutes of Health Awards CA-16042 and AI-28697, the Jonsson Cancer Center, the UCLA AIDS Institute and the UCLA School of Medicine.
Median-effect analysis. The Chou and Talalay median-effect principle was used to evaluate drug interaction in drug combination studies from cell viability assays (27). Median effect doses (Dm; analogous to the IC50) were determined from dose response experiments using individual inhibitors (LY94002 or Akti-1/2) or chemotherapeutic agents (cisplatin, paclitaxel, gemcitabine or topotecan). Ratios of the Dm's for combinations of a particular inhibitor (e.g. LY294002) and a particular chemotherapeutic agent (e.g. cisplatin) were then determined. The drug combinations were applied at a fixed ratio over a range of concentrations. Interaction (synergy, additivity or synergism) between pairs of drugs was determined based on the multiple drug effect equation of Chou and Talalay and was quantified by the combination index (CI) (27). The CI indicates synergism when = <0.9, additivity when = 0.9-1.1 and antagonism when = >1.1. The Fa (fraction affected)-CI plot indicates the expected degree of drug interaction and the corresponding Fa based on experimental data. Calculations were based on the mutually exclusive assumption of the mode of activity of the drugs. Dose effect curves, CIs and Fa-CI plots were generated in CalcuSyn (Biosoft, Ferguson, MO, USA).
Statistical analysis. All the statistical analyses were performed using GraphPad Prism, Version 4.00c for Macintosh (GraphPad Software, www.graphpad.com). Two-tailed unpaired t-tests were used to calculate the significance of differences between cell cycle phases from the various treatment conditions in the cell cycle flow cytometric analyses. Treatment with inhibitor or chemotherapeutic agent alone was compared to untreated control cells. Combination treatment with chemotherapeutic agent and inhibitor was compared to single treatment with the respective chemotherapeutic agent.
Results
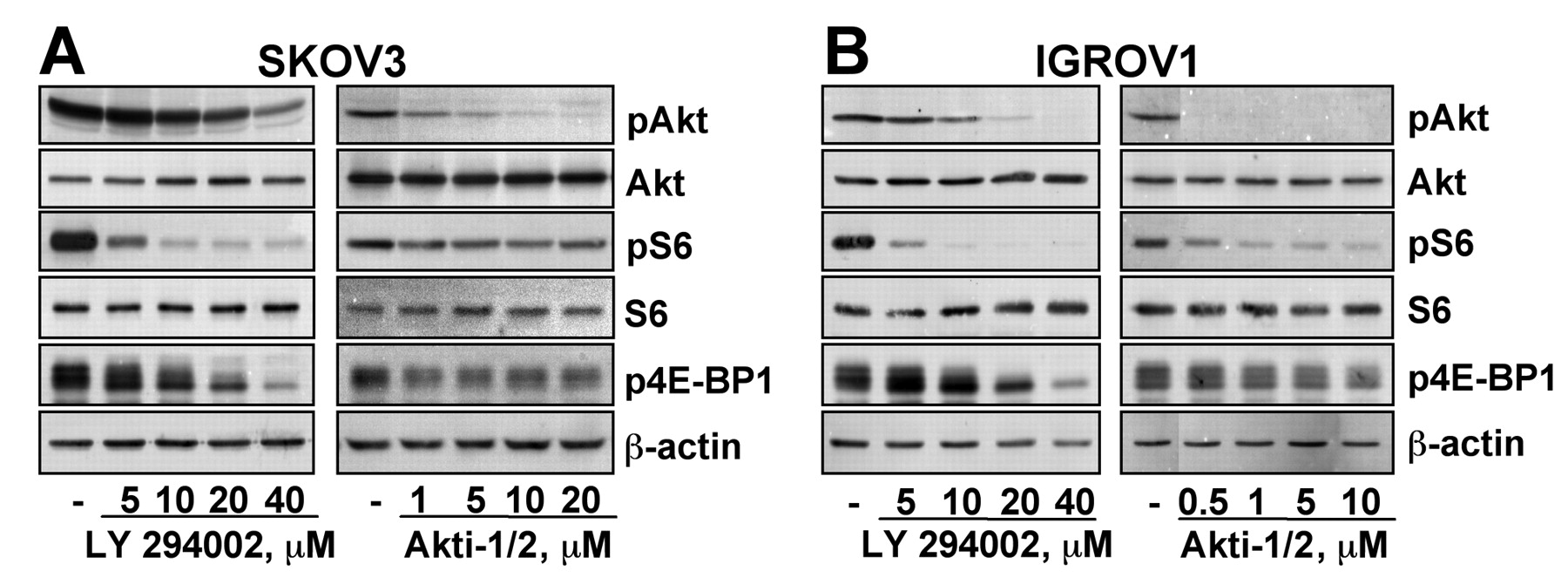
Effect of LY294002 and Akti-1/2 on PI3K/Akt pathway signaling. Treatment of the SKOV3 (Figure 1A) and IGROV1 (Figure 1B) cells with LY294002 or Akti-1/2 decreased phosphorylation of Akt (pAkt), S6 (pS6) and 4E-BP1 (p4E-BP1) in a dose-dependent manner in both cell lines. The levels of total Akt, S6 and β-actin are shown as loading controls.

Effect of LY294002 and Akti-1/2 on phosphorylation of Akt, S6 and 4E-BP1 in SKOV3 and IGROV1 cells. SKOV3 (A) and IGROV1 (B) cells were serum-starved overnight, followed by treatment with LY294002 or Akti-1/2 for 24 h. Total cell extracts were analyzed by immunoblotting for pAkt, pS6 or p4E-BP1. Total Akt, S6 and β-actin levels are shown as loading controls. Representative blots of 3 independent experiments are shown.
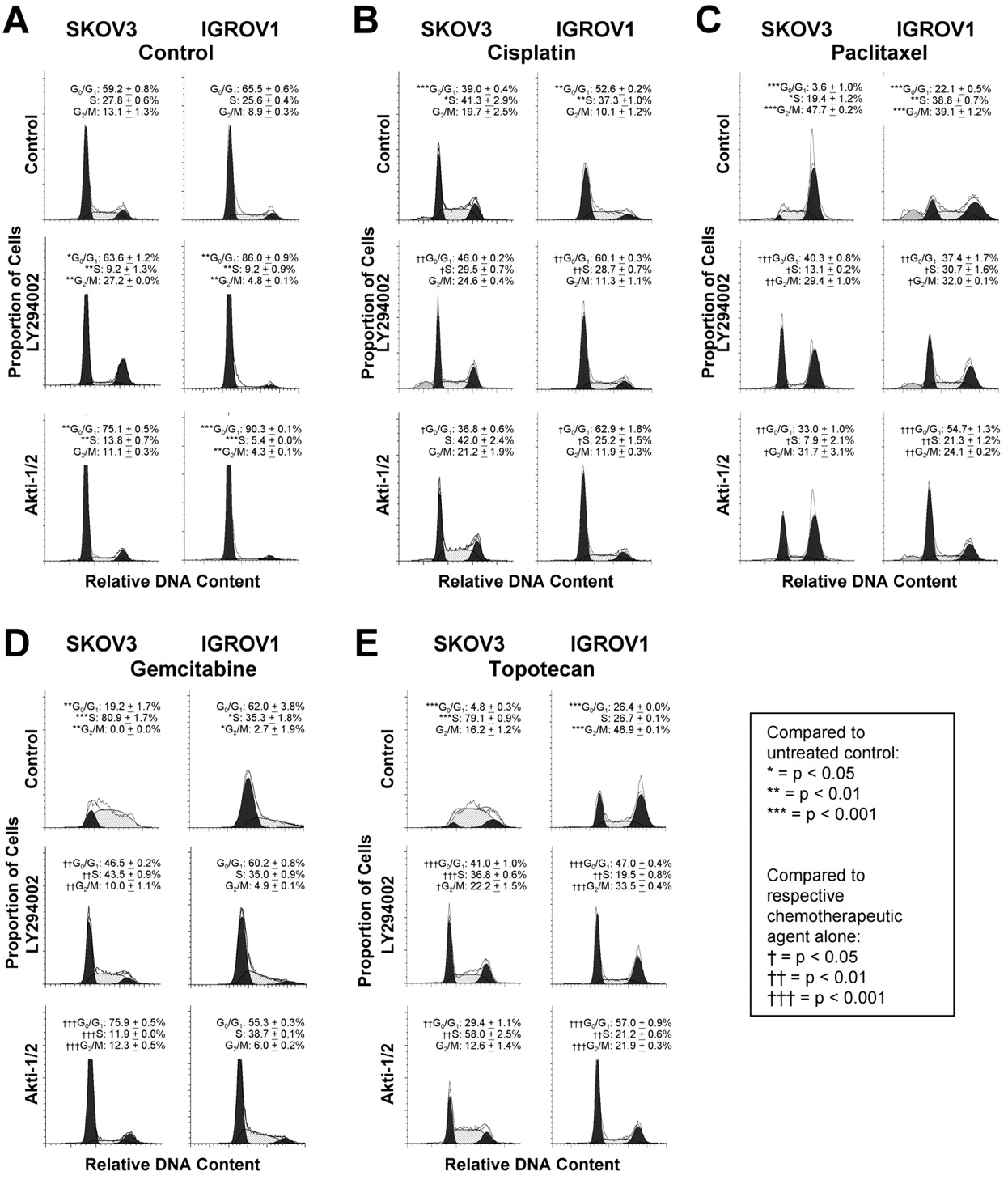
Effect of PI3K/Akt pathway inhibition on cisplatin, paclitaxel, gemcitabine or topotecan-treated cells. The cell cycle distribution of untreated SKOV3 cells showed 59.2±0.8% of the cells in G0/G1, 27.8±0.6% in S and 13.1±1.3% in G2/M (Figure 2A left). For IGROV1 cells under control conditions, 65.5±0.6% of the cells were found to be in G0/G1, 25.6±0.4% in S, and 8.9±0.3% in G2/M (Figure 2A right). Exposure of the cells to LY294002 or Akti-1/2 increased the proportion of cells in G0/G1 and decreased those in S-phase (Figure 2A).
Cisplatin treatment alone increased the cells in S-phase (SKOV3: 41.3±2.9%; IGROV1: 37.3±1.0%) and decreased the percentage of SKOV3 and IGROV1 cells in the G0/G1-phase (SKOV3: 39.0±0.4%; IGROV1: 52.6±0.2%) compared to untreated cells (Figure 2B). However, when the cells were treated with a combination of LY294002 and cisplatin, the percentage of cells in G0/G1 was higher than the one in the cisplatin alone treated cells (SKOV3: 46.0±0.2%; IGROV1: 60.1±0.3%). In contrast, the number of cells in S-phase decreased with combined LY294002 and cisplatin treatment compared to cisplatin treatment alone. Similarly, in IGROV1 cells treatment with Akti-1/2 reversed the cisplatin-induced decrease of cells in G0/G1 and increase of cells in S-phase. Paclitaxel treatment caused an accumulation of SKOV3 and IGROV1 cells in G2/M (SKOV3: 47.7±0.2%; IGROV1: 39.1±1.2%) compared to untreated cells, while only 3.6%±1.0% and 22.1±0.5%, respectively, remained in G0/G1 (Figure 2C). When combined with LY294002, the percentage of cells in G2/M decreased, while the cells in G0/G1 increased significantly (SKOV3: 40.3±0.8%; IGROV1: 37.4±1.7%) compared to paclitaxel alone. The reversal of paclitaxel-induced G2/M accumulation with PI3K/Akt inhibition was only partial as the percentage of cells in G2/M still remained higher than in untreated cells. Combination treatment with paclitaxel and Akti-1/2 had similar effects.
In the SKOV3 cells, gemcitabine increased the proportion of cells in the S phase of the cell cycle (80.9±1.7%), while the majority of the remaining cells were found to be in G0/G1 (19.2±1.7%) compared to untreated control cells (Figure 2D left). Upon addition of LY294002 and gemcitabine, the percentage of cells in the S-phase decreased with a shift towards the G0/G1 phase (43.5±0.9%). The shift of cells from the S-phase to G0/G1 was even more pronounced when gemcitabine was combined with Akti-1/2 (75.9±0.5%). In the IGROV1 cells, gemcitabine had only minor effects with 62.0±3.8% of cells still remaining in G0/G1 (Figure 2D right). Accordingly, no significant changes were observed in the combination treatments with gemcitabine and LY294002 or Akti-1/2.
Treatment of the SKOV3 cells with the topoisomerase I inhibitor topotecan increased the percentage of cells in S-phase (79.1±0.9%) compared to untreated control (Figure 2E left), while only 4.8±0.3% of the cells remained in G0/G1. However, the combination of topotecan and LY294002 showed a significant reduction in the percentage of cells in S-phase, while the percentage of cells in G0/G1 increased to 41.0±1.0%. Akti-1/2 treatment showed a similar reversal of topotecan-induced cell cycle shifts. Topotecan treatment of the IGROV1 cells increased the percentage of cells in G2/M (46.9±0.1%) compared to untreated control, with a corresponding decrease in G0/G1 (26.4±0.0%) (Figure 2E right). Combination treatment with LY294002 or Akti-1/2 reversed this effect by shifting cells to the G0/G1-phase (LY294002: 47.0±0.4%; Akti-1/2: 57.0±0.9%) compared to topotecan alone.
Effect of PI3K/Akt pathway inhibition on the cytotoxic effects of cisplatin and paclitaxel, gemcitabine and topotecan. Based on the multiple drug effect equation of Chou and Talalay, LY294002 enhanced the cytotoxic effect of cisplatin in a synergistic manner in both the SKOV3 and IGROV1 cells. The combination index at ED75 (CI75) for the SKOV3 cells was 0.42 and 0.30 for the IGROV1 cells (Figure 3A). Combination treatment with cisplatin and Akti-1/2 showed a similar synergistic effect (SKOV3: CI75=0.71; IGROV1: CI75=0.19).
LY294002 also augmented the paclitaxel-induced decrease in cell proliferation in a synergistic manner (SKOV3: CI75=0.79; IGROV1: CI75=0.60) (Figure 3B). The enhanced effect was additive with Akti-1/2 (SKOV3: CI75=0.97; IGROV1: CI75=0.97).
In contrast, PI3K/Akt pathway inhibition antagonized, rather than synergized, the effects of gemcitabine in both cell lines. In the SKOV3 cells, the CI75 for the combination of gemcitabine and LY294002 or Akti-1/2 was 1.64 and 4.24, respectively (Figure 3C). Similarly, in the IGROV1 cells gemcitabine combined with LY294002 yielded a CI75 of 27.01, and 1.55 for the combination with Akti-1/2. Likewise, LY294002 and Akti-1/2 antagonized the effects of topotecan in the SKOV3 cells (LY294002: CI75=1.67; Akti-1/2: CI75=1.39) (Figure 3D). A lack of synergy was observed in the IGROV1 cells when treated with topotecan and LY294002 (CI75=1.02) or Akti-1/2 (CI75=1.12).
Discussion
The hypothesis that PI3K/Akt pathway inhibition-induced cell cycle arrest in G0/G1 can modulate the cytotoxic effects of certain chemotherapeutic agents commonly used for the treatment of ovarian and other carcinomas was investigated. Antagonization of cytotoxic chemotherapy was demonstrated by PI3K/Akt pathway inhibition when human ovarian cancer cells were treated with a combination of LY294002 or Akti-1/2 and gemcitabine or topotecan. Both chemotherapeutic agents exert their main effects during the S and G2-phases of the cell cycle (23). Treatment of the ovarian cancer cells with gemcitabine or topotecan alone caused cell cycle arrest in S-phase. However, when the cells were treated with either agent and concomitant PI3K/Akt pathway inhibition, S-phase accumulation was reversed and the cells were shifted to the G0/G1-phase. In the cell proliferation assays Chou and Talalay median-effect principle analysis demonstrated an antagonistic effect of LY294002 and Akti-1/2 on the effects of gemcitabine and topotecan. These observations suggest that G0/G1 arrest induced by LY294002 and Akti-1/2 precluded cells from progressing into the S and G2-phases, and thus prevented gemcitabine and topotecan from exerting their cytotoxic effects.
Since paclitaxel is mainly effective in the M-phase of the cell cycle (23), PI3K/Akt pathway inhibition was also expected to antagonize its effects on cell proliferation. However, LY294002 and Akti-1/2 enhanced the cytotoxic effects of paclitaxel in a synergistic manner. Various mechanisms could contribute to this observed synergy. Although LY294002 and Akti-1/2 reversed paclitaxel-induced M-phase accumulation, this reversal was not complete and possibly suggests that a sufficient number of cells were able to progress to the M-phase even in the presence of the PI3K/Akt pathway inhibitors. Moreover, in response to paclitaxel, cancer cells can activate the PI3K/Akt pathway resulting in, for example, phosphorylation and inactivation of the proapoptotic Bcl-2 family member Bcl-associated agonist of cell death (BAD) (28). PI3K or Akt inhibition prevents the activation of this important survival mechanism, which may explain the synergistic effect of the inhibitors on paclitaxel-induced cell proliferation. Other studies have previously reported that PI3K/Akt pathway inhibition can enhance paclitaxel effects (29-31).
Similarly, although PI3K/Akt pathway inhibition also arrested cisplatin treated cells in G0/G1, in the cell proliferation assays LY294002 and Akti-1/2 enhanced the cytotoxic effects of cisplatin in a synergistic manner. Various cisplatin related effects might contribute to this synergy. Cisplatin is a DNA alkylating agent that crosslinks DNA and elicits its effects in a rather cell cycle unspecific manner (23). In addition, similar to paclitaxel, cisplatin can activate PI3K/Akt signaling as a cellular survival mechanism (32-34). Various other studies have demonstrated a similar effect of PI3K/Akt pathway inhibition on cisplatin-induced effects (33, 35-38).
The data presented in this study might have important implications for the design of clinical trials using PI3K/Akt pathway inhibition in combination with chemotherapeutic agents. Inhibition of this growth-promoting and apoptosis-inhibiting pathway has been proposed as a strategy to sensitize tumors to chemotherapy or to reduce or delay the development of chemoresistance (19, 20). PI3K/Akt pathway inhibitors are being studied in clinical trials for various malignancies including ovarian cancer (39-42). It is currently unclear which patients will benefit from treatment using PI3K/Akt inhibitors, and whether combination with a standard chemotherapeutic agent would be more effective. Based on the present data, chemotherapeutics that are primarily effective in the S or G2-phase such as gemcitabine or topotecan might not be the ideal agents due to PI3K/Akt pathway inhibition-induced G1 cell cycle arrest.
In summary, cell cycle analyses demonstrated that the PI3K/Akt pathway inhibitors LY294002 or Akti-1/2 arrested cells in G1 phase, and furthermore reversed the cell cycle effects of cisplatin, paclitaxel, gemcitabine and topotecan when cells were treated in combination. In cell proliferation studies, the inhibitors synergistically enhanced the cytotoxic effects of cisplatin and paclitaxel, but failed to sensitize cells to gemcitabine or topotecan.

Effect of LY294002 and Akti-1/2 on cisplatin, paclitaxel, gemcitabine and topotecan-treated SKOV3 and IGROV1 cells. SKOV3 and IGROV1 cells were treated with LY294002 or Akti-1/2 alone (A), or cisplatin (B), paclitaxel (C), gemcitabine (D) or topotecan (E) in the absence or presence of inhibitor. Cell cycle distribution was quantified by flow cytometric analysis of propidium iodide-stained cells. Concentrations used: SKOV3: LY294002, 20 μM; Akti-1/2, 5 μM; cisplatin, 20 μg/ml; paclitaxel, 10 nM; gemcitabine, 100 nM; topotecan, 100 nM. IGROV1: LY294002, 10 μM; Akti-1/2, 5 μM; cisplatin, 5 μg/ml; paclitaxel, 10 nM; gemcitabine, 100 nM; topotecan, 250 nM. Results shown are representative of 3 or more independent experiments performed in duplicate.
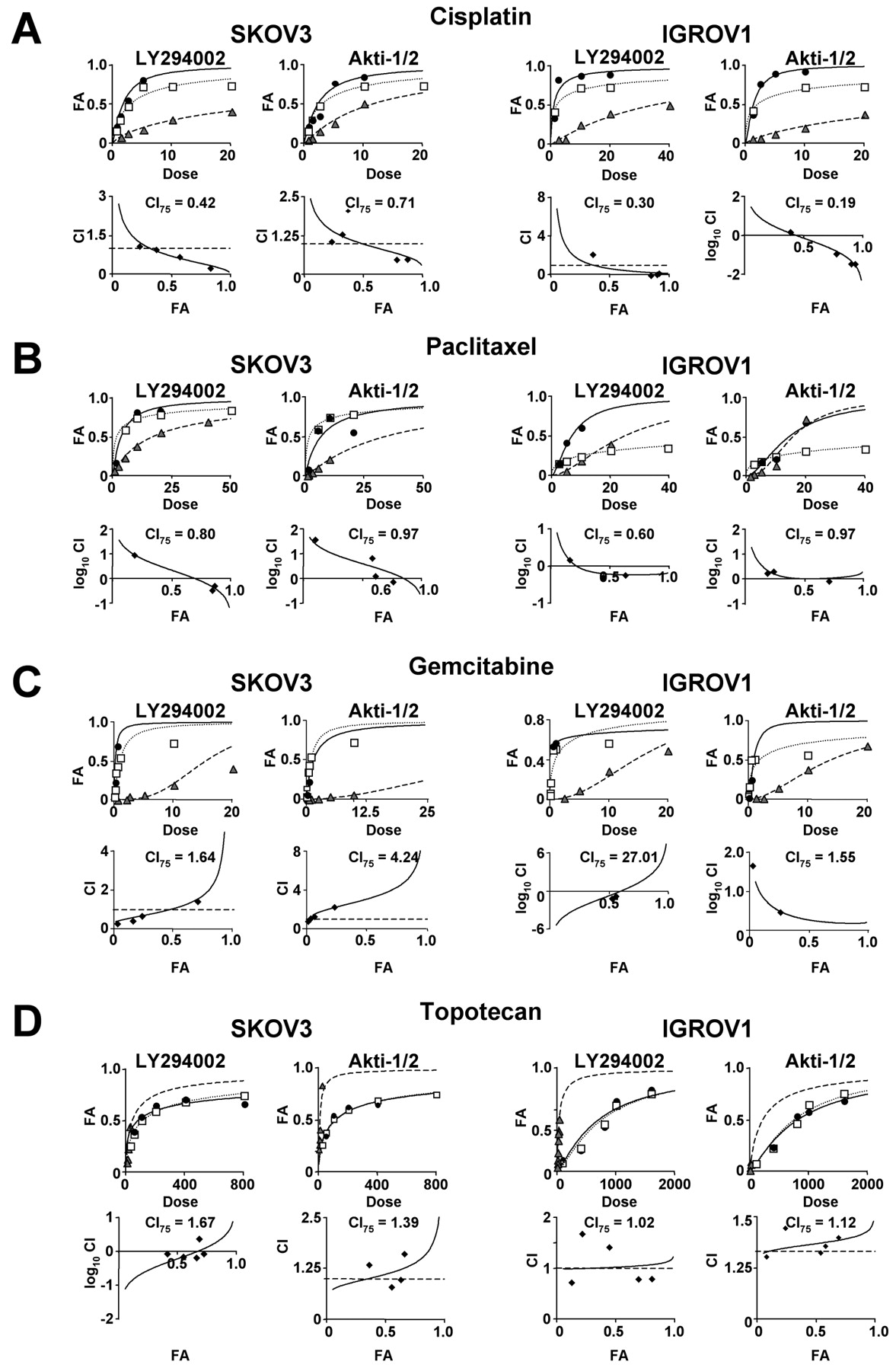
Effect of LY294002 and Akti-1/2 on the cytotoxic effects of cisplatin and paclitaxel, gemcitabine and topotecan. SKOV3 and IGROV1 cells were treated with LY294002 (μM) or Akti-1/2 (μM) alone, or cisplatin (μg/ml) (A), paclitaxel (nM) (B), gemcitabine (nM) (C) or topotecan (nM) (D) alone or with the combination of inhibitor and chemotherapeutic agent. Cell proliferation was determined via the XTT cell viability assay. The upper panel shows the dose response curve of single and combination drug treatment. The lower panel shows the Fa-CI curve. The CI at ED75 is indicated. Experiments were performed in quadruplicate or more, and results shown are representative of 2 or more independent experiments.  , inhibitor alone; □, chemotherapeutic agent alone; ●, inhibitor + chemotherapeutic agent.
, inhibitor alone; □, chemotherapeutic agent alone; ●, inhibitor + chemotherapeutic agent.
Acknowledgements
This study was supported by the Ovarian Cancer Research Foundation/Liz Tilberis Scholarship, the Gynecologic Cancer Foundation/Florence and Marshall Schwid Ovarian Cancer Award, the STOP Cancer Career Development Award, and the NIH Women's Reproductive Health Research (WRHR) Career Development Program (2K12HD001281). C.S. was supported by a NIH T32 training grant (CA009056).
Footnotes
-
↵* These Authors contributed equally to this study.
- Received November 21, 2011.
- Revision received December 30, 2011.
- Accepted January 3, 2012.
- Copyright© 2012 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Systematic characterization of therapeutic vulnerabilities in Multiple Myeloma with Amp1q reveals increased sensitivity to the combination of MCL1 and PI3K inhibitors
- FOXO1 mitigates the SMAD3/FOXL2C134W Transcriptomic Effect in a Model of Human Adult Granulosa Cell Tumor
- Targeting AKT elicits tumor suppressive functions of FOXO transcription factors and GSK3 kinase in Multiple Myeloma
- A First-in-Human, Phase I, Dose-Escalation Study of TAK-117, a Selective PI3K{alpha} Isoform Inhibitor, in Patients with Advanced Solid Malignancies