


Abstract
Polo-like kinase 1 (PLK1) is the master regulator of mitosis and a target for anticancer therapy. To develop a marker of PLK1 activity in cells and tumour tissues, this study focused on translational controlled tumour protein (TCTP) and identified serine 46 as a site phosphorylated by PLK1 in vitro. Using an antibody raised against phospho-TCTP-Ser46, it was demonstrated that phosphorylation at this site correlates with PLK1 level and kinase activity in cells. Moreover, PLK1 depletion by siRNA or inactivation by specific inhibitors caused a correspondent decrease in phospho-TCTP-Ser46 signal validating this site as a direct marker of PLK1. Using this marker, the study characterized PLK1 inhibitors in cells by setting up a high-content assay and finally immunohistochemical assay suitable for following inhibitor activity in preclinical tumour models and possibly in clinical studies was developed.
The Polo-like kinase (PLK) family includes four members (PLK1, PLK2, PLK3 and PLK4) in humans, all having in common a conserved C-terminal sequence denoted as the Polo-box domain (1). PLK1 is the most well-characterised member of the family, PLK2 and PLK3 show the highest degree of homology to PLK1 with similar Polo-box and kinase domains, whereas PLK4 contains only partial Polo-box domain and is the more distant member of the family. PLK1 is considered the master regulator of all mitosis steps as reviewed in (2), while the roles of the other members of the family have not yet been completely elucidated. PLK1 constitutive expression in NIH3T3 cells induces reduced serum requirements and growth in soft agar. In addition, PLK1 in vitro transformed cells form tumours after subcutaneous injection in athymic nude mice (3). Furthermore, PLK1 depletion (4) or inhibition by small molecules is sufficient to drive a mitotic cell-cycle block followed by apoptosis in tumour cell lines and xenograft models. PLK1 is present in all proliferating normal tissues, while it is absent in non-proliferating ones. It is overexpressed in different tumour types including lung, colon, prostate, ovary, breast, head and neck squamous cell carcinoma, melanoma and its overexpression often correlates with poor prognosis. All of these observations indicate PLK1 as a promising target for anticancer therapy (5, 6) and small molecules that inhibit its activity are currently undergoing clinical trials (7).
Although many different substrates of PLK1 have been described in the literature, including cdc25c (8), cohesins (9), cyclinB1 (10), Wee1 (11), Myt1 (12), P53 (13) and Emi1 (14), at present no reliable direct marker for evaluation of PLK1 activity in cells and tissues has been reported, possibly due to the low stoichiometry of phosphorylation, the low abundance or the degradation after phosphorylation of the substrate proteins. The identification of these substrates in literature has been performed by overexpression of the substrate itself or by immunoprecipitation studies. However, these methods are barely suitable for marker evaluation in animal models and patients. Thus, at present, clinical studies with PLK1 inhibitors are not supported by a pharmacodynamic audit trail from tumour tissues (15) due to the fact that a direct marker of PLK1 activity is not yet available. PLK1 inhibition or depletion in cells induce a mitotic block that is usually measured by cytofluorimetric analysis or by evaluating the levels of histone H3 phosphorylated on serine 10. Unfortunately, mitotic marker increase, although clearly related to PLK1 depletion or inhibition, is not a direct effect and is also induced by compounds that do not interact directly with PLK1, such as microtubule stabilisers or destabilisers or inhibitors of mitotic kinesins.
With the aim of identifying a substrate suitable to follow PLK1 activity in cells and possibly in xenograft tumours, the present study characterised the phosphorylation of translationally controlled tumour protein (TCTP) by PLK1. TCTP is a highly conserved protein identified in all eukaryotic organisms. TCTP was initially identified in a two-hybrid screening as an interactor of the Polo-box domain of PLK1 and was then demonstrated to be an in vitro substrate for recombinant PLK1 (16). Mutational analysis has identified serine 46 and serine 64 as residues phosphorylated specifically by PLK1 on TCTP (16). In addition, using mass spectroscopy, a recent publication confirmed that PLK1 phosphorylates in vitro recombinant TCTP on serine 46 (17).
Using mass spectroscopy, the present study confirmed serine 46 as the main residue phosphorylated by PLK1 in TCTP and identified threonine 65 as a new secondary phosphorylation site. An antibody was raised against TCTP phosphorylated on serine 46 (phospho-TCTP-Ser46) and was demonstrated to be highly specific both in vitro and in cells. Furthermore, the antibody detected the expression of phosphorylated TCTP in xenograft tumour tissues by immunohistochemistry. This antibody was used to set up a cellular assay appropriate for rapidly ranking the potency of many small-molecule inhibitors of PLK1. Thus, using this antibody, it was possible to show that phospho-TCTP-Ser46 may be used as a suitable direct marker of PLK1 inhibition not only in cells but also in future in vivo experiments.
Materials and Methods
Cloning, expression and purification of recombinant TCTP. Full-length TCTP was amplified from human foetal brain cDNA library by PCR and cloned in a vector for bacterial expression. For purification purposes, an N-terminal His-GST tag and a PreScission® (Little Chalfont, Buckinghamshire, UK) protease cleavage site were added. His-GST-TCTP was expressed in Escherichia coli BL21 (DE3) strain from Merck-Novagen (Darmstadt, Germany). E.coli transformed cells were grown at 37°C to an optical density at 600 nm (OD600nm) of 0.6 and protein expression was induced with 0.5mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) from Sigma-Aldrich (St Louis, MO, USA) at 21°C. Cells were collected after 24-h incubation. The cellular pellet was lysed in 50 mM Tris-HCl buffer pH 7.4, 150 mM NaCl, 20 mM DTT and purified by affinity chromatography on Glutathione Sepharose from GE Healthcare. Elution was performed by PreScission® protease cleavage of the GST tag.
Cloning, expression and purification of recombinant PLK1 kinase domain. PLK1 kinase domain (corresponding to residues 2-345) was amplified by PCR from a human testis cDNA library. For cloning purposes, the oligonucleotides included attB sites in order to obtain an attB-flanked PCR product suitable for cloning using the Gateway® system (Invitrogen, Carlsbad, CA, USA). For purification purposes, the forward primer included a PreScission® protease cleavage site. The resulting PCR product was cloned into the baculovirus expression vector pVL1393 (Invitrogen) Gateway®-modified. For expression and purification purposes, a GST tag was added to N-terminal PLK1 kinase domain. Baculoviruses were generated by cotransfecting Sf9 insect cells with the expression vector and the viral DNA using the BaculoGold® transfection kit (BD Biosciences, Pharmingen, San Diego, CA, USA). After 48 h of infection, cells were recovered and pellets were resuspended in lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.2% CHAPS, 20 mM DTT, 10% glycerol and protease inhibitors) and lysed by sonication. Lysates were cleared by centrifugation at 24000×g for 30 min and loaded on a Glutathione Sepharose 4FF® (GE Healthcare) column. After extensive washes, recombinant protein was cleaved and eluted by incubation with PreScission® protease.
Gel kinase assay. For in vitro kinase assays, 10 μM recombinant TCTP was incubated for 30 min at 37°C in the presence of 50 mM HEPES pH 7.5, 10 mM MgCl2, 2 mM DTT, 50 μM ATP and 20 nM PLK1 kinase domain. For liquid chromatography/mass spectrometry analysis, the enzyme to substrate ratio was increased to 1:50 and the ATP concentration was 500 μM.
Liquid chromatography / Mass spectrometry (LC/MS) analysis. A total of 20 μg of TCTP protein was analysed before or after phosphorylation with PLK1. The chromatographic separations were performed on an Agilent 1100 (Agilent Technologies, Santa Clara, CA, USA) instrument using a C-4 column (Grace-Vydac, Deerfield, IL, USA) (internal diameter, 2.1 mm; length, 25 cm; particle size, 5 μm; pore size, 300 Å). The eluate from the column was sent directly to the MS instrument. Positive ion ESI mass spectra were obtained using 1946 single quadrupole mass spectrometer (Agilent) with an orthogonal ESI source. The needle voltage was set at 3000 V the mass range was set to m/z 600-2000. The molecular weight of the protein was obtained by deconvolution of the ESI mass spectrum using the Chemstation Deconvolution software (Agilent).
Enzymatic digestion. TCTP digestion was performed with V8 protease in a buffer containing 50 mM ammonium bicarbonate. A total of 10 μg of phosphorylated or non-phosphorylated TCTP was incubated with 1 μg of V8 protease for 3 h at 37°C. Sample desalting was performed by ZipTip C18 (Millipore, Billerica, MA, USA) using a standard protocol.
MALDI-MS. Peptides for matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF-MS) analysis were eluted from ZipTip C18 resin (Millipore) with 5 μl 60% v/v acetonitrile. A volume of 0.5 μl of this sample was spotted on a MALDI plate with 0.5 μl of α-cyano-4-hydroxycinnamic acid (10 mg/ml in 50:50 acetonitrile: water containing 0.1% trifluoroacetic acid) and analysed on a Voyager DE-PRO instrument (Applied Biosystems, Foster City, CA, USA). All spectra were collected in reflector mode using four peptides of known mass as external calibration standards.
Nano-ESI-MS/MS. Tandem MS was performed on a hybrid quadrupole-time of flight instrument (Q-ToF2; Waters, Milford, MA, USA) equipped with a Z-spray source and calibrated by injecting a solution of Glu-Fibrinopeptide (Sigma-Aldrich) (0.5 pM/μl) at 0.5 μl/min, applying 3.2 kV to the spraying capillary and a collision energy of 29 V. For phosphosite assignment by MS/MS, the desalted samples were dried down in a speed vacuum, redissolved in a 1:1 acetonitrile/0.2% formic acid mixture and loaded directly into nanoflow probe tips. Each fraction was subjected to MS analysis over the 100-2000 m/z scan range; MS/MS analyses were performed by manually selecting the phosphorylated peptides and fragmenting them under user-defined parameters of collision energy.
Cell culture. A2780, human ovarian cancer cells (European Collection of Cell Culture, Colindale, London, UK), were maintained in RPMI medium; U2OS, human osteosarcoma (American Type Culture Collection, Manassas, VA, USA), and HCT-116, human colon carcinoma cells (European Collection of Cell Culture), were maintained in McCoy's medium. All cell lines were supplemented with 10% foetal calf serum (FCS) and 2 mM L-glutamine and cells were cultured at 37°C in a humidified incubator in the presence of 5% CO2. The cells were treated with PLK1 inhibitors at the indicated concentrations and times, starting from a stock solution of 10 mM in DMSO. DMSO alone was used as negative control, nocodazole was used at 75 ng/ml for 18 h, thymidine was used at 2 mM for 20 h and MG132 was used at 5 μM for 1 h (all from Sigma-Aldrich).
Flow cytometric analysis of DNA content. Fixed cells in 70% ethanol/PBS were washed with PBS to remove fixative and stained with 25 μg/ml propidium iodide, 5 μg/ml RNase and 0.125 μg/ml Nonidet P40. Cells were kept at room temperature for 60 min and analysed by a BD FACSCalibur™ system (BD Biosciences, San Jose, CA, USA).
Immunoblotting. Cells were lysed in 125 mM Tris-HCl (pH 6.8) and 2% SDS and heated for ten minutes at 95°C. Total proteins (20 μg), as determined by BCA protein assay (Pierce, Rockford, IL, USA), were separated by 4-12% SDS-PAGE. Immunoblot analysis was done using the following antibodies: anti-PLK1 (Zymed, San Francisco, CA, USA), anti-phospho-TCTP-Ser46 (custom made by Zymed), anti-TCTP (BD Transduction Laboratories, Lexington, KY, USA), anti-phospho-histone H3-Ser10 (Upstate Biotechnology, Waltham, MA, USA), anti-histone H3 (Abcam Cambridge, MA, USA), anti-phospho-NPM-Thr199 (Cell Signaling, Danvers, MA, USA) and cyclin B1 (BD Transduction Laboratories). Immunoblot analysis was performed using standard methods. SuperSignal chemiluminescence kit (Pierce) was used for detection.
Immunofluorescence of cells. U2OS cells were fixed with 3.7% paraformaldehyde and permeabilised with 0.5% Triton ×100. Saturation was performed with 3% bovine serum albumin (BSA). Primary antibodies were used at the following dilutions: anti-phospho-TCTP-Ser46 1:100 and TCTP 1:100. As secondary antibody, goat anti-rabbit-Cy3 and anti-mouse-Cy2 conjugates were used (GE Healthcare). DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich).
Immunohistochemistry and immunofluorescence of xenograft tumours. A2780 cells were transplanted subcutaneously in female Hsd, athymic nu/nu mice (Harlan) aged five to six weeks. All procedures adopted for housing and handling of animals were in strict compliance with the Italian Legislative decree N 116 dated January 27, 1992, and the European Communities Council Directive N86/609 EEC concerning the protection of animals used for experimental and other scientific purposes and according to institutional policy regarding the care and use of laboratory animals. Mice bearing a palpable tumour (400-600 mm3) were treated once intravenously with vehicle or paclitaxel (BMS, New York City, NY, USA) at 30 mg/kg dose. After 24 h, tumours were harvested, formalin-fixed and paraffin-embedded. Paraffin sections (3-μm thick) were deparaffinised in xylene, rehydrated in graded ethanol and transferred to PBS. Sections were placed in citrate buffer (pH 6.0) and thermally processed with two cycles in an autoclave 2100 Retriever (PickCell Laboratories, Amsterdam, the Netherlands). After washing with PBS, endogenous peroxidase was blocked using 3% hydrogen peroxide in PBS for 10 min. The tissues were washed with PBS plus 0.01% Tween 20 and incubated for 30 min at room temperature with a protein-blocking solution consisting of PBS, containing 10% normal goat serum and 0.01% Tween 20. The sections were incubated with polyclonal antibodies against phospho-TCTP-Ser46 and phospho-histone H3-Ser10 diluted in blocking solution for one hour at 37°C and room temperature, respectively. The samples were rinsed twice with PBS plus 0.01% Tween 20, incubated for 30 min at room temperature with an EnVision system HRP anti-rabbit and DAB substrate chromogen (Dako, Golstrup, Denmark) for immunohistochemistry and goat anti-rabbit-Cy3 conjugates (GE Healthcare) for immunofluorescence.
RNA interference. siRNA oligonucleotides (Dharmacon, Lafayette, CO, USA) were synthesised toward the following sequence (sense strand): PLK1 (5′-CGAGCUGCUUAAUGACGAG-3′). Transfections were performed using Oligofectamine (Invitrogen) according to the manufacturer's recommendations. siRNAs were used at a final concentration of 60 nM.
Phospho-TCTP-Ser46 array scan assay. An array scan assay for phospho-TCTP-Ser46 studies was performed on U2OS osteosarcoma cells seeded at 5000 cells/well in 96-well plates incubated for 24 h at 37°C. The cells were then synchronised in M phase by 75 ng/ml nocodazole treatment for 18 h. The medium was then replaced with fresh medium containing test compounds at the required concentrations. Concentration curves were prepared in DMEM plus 10% FCS starting from compound stock solutions in DMSO with final DMSO concentration 0.1% (v/v). Duplicate wells for each concentration point were prepared, with a maximal compound concentration of 10 μM. After addition of compounds, the plates were incubated for 1 h at 37°C. The cells were fixed in 3.7% paraformaldehyde solution and then stained with anti-phospho-TCTP-Ser46 antibody in 1% (w/v) BSA. Cy2-anti-rabbit antibody (GE Healthcare) was then used as secondary antibody. In parallel, 1 μg/ml of DAPI (Sigma-Aldrich) was added to detect nuclear staining. Fluorescent staining was assessed by ArrayScan vTi instrument (Cellomics/Thermo Scientific, Waltham, MA, USA), with a 10×0.5 N.A. objective (Zeiss, Jena, Germany), and applying the Nuclear translocation V3 algorithm (Cellomics/Thermo Scientific), with a XF100 filter. At least ten fields, corresponding to at least 900 cells, were read for each well. IC50 values were calculated, representing the compound concentration at which the cellular signal was diminished by 50% compared to synchronised control cells.
Phospho-histone H3-Ser10 and cyclin B array scan assay. The assays were performed on U2OS cells growing asynchronously using the same conditions for treatment with compounds reported for the phospho-TCTP-Ser46 array scan. Primary antibodies were mouse monoclonalphospho-histone H3-Ser10 (Cell Signaling) and rabbit polyclonal cyclin B1 (Santa Cruz). Cy2-conjugated goat anti-mouse and anti-rabbit secondary antibodies were from GE Healthcare.

TCTP was phosphorylated by PLK1 and mono- and di-phosphorylated forms were identified. A: Phosphorylation of recombinant TCTP and α-casein by PLK1 kinase domain expressed in insect cells was evaluated by gel kinase assay. Clear phosphorylation of TCTP was observed with a level comparable to or even higher than that of the generic substrate α-Casein. B: LC/MS analysis of non-phosphorylated TCTP and TCTP phosphorylated by PLK1. After phosphorylation, a complete conversion of non-phosphorylated TCTP (0P) to mono- (1P) and diphosphorylated (2P) forms was observed.
Biochemical kinase assay IC50 and inhibition of cell proliferation. Biochemical IC50 and inhibition of cell proliferation were performed as previously reported (18).
Results
TCTP in vitro phosphorylation by PLK1 and identification of phosphosites. To verify that TCTP is a direct substrate of PLK1, the full-length human TCTP expressed in E coli was phosphorylated in vitro with PLK1 and the phosphorylated protein was assessed using a gel kinase assay. As control for kinase activity, α-casein was used, which is a generic substrate for PLK1. The levels of 32P incorporation in TCTP were comparable or even higher than those for α-casein after a short reaction time, indicating that TCTP is efficiently phosphorylated by PLK1 (Figure 1A).
Subsequently, the level of TCTP phosphorylation was evaluated quantitatively by mass spectrometry and the analysis revealed two phosphorylation sites with approximately 55% of monophosphorylated and 45% of diphosphorylated protein (Figure 1B), in agreement with published results (16). More in-depth characterization of the phosphorylation sites was carried out using a standard phosphopeptide mapping approach by the enzymatic digestion of the full-length phosphoprotein with V8 protease and the analysis of the resulting peptides by MALDI-TOF/MS. This analysis revealed the presence of three peaks matching the molecular weight of phosphorylated peptides corresponding to the regions 33-63 (3157 Da), 59-86 (3139.5) and 64-80 (1972.9 Da) of TCTP. The phosphorylation of the peptides was confirmed through alkaline phosphatase treatment as a clear shift of 80 Da corresponding to the lack of a phosphate group observed in phosphatase-treated samples (Figure 2A-D).
Phosphorylated peptides were selected for the MS/MS analysis in order to identify the phosphorylated residues. The fragmentation of the peptide corresponding to the peak at m/z 3157.4 identified a residue of phospho-serine at the position 46 in the sequence of TCTP, in agreement with published data (16, 17). The same analysis performed on the ion at m/z 1972.9 indicated that the phosphate group is located on the dipeptide Ser64-Thr65. Surprisingly, further analysis revealed the presence of a dehydroamino butyric acid residue (Dhb) signal in the MS/MS spectrum, a known degradation product of phospho-Thr after the loss of phosphoric acid during fragmentation. From these data and in contrast to (16), it is concluded that the phosphate group is located on Thr65 instead of Ser64. Whereas the Ser46 site appeared to be entirely phosphorylated, a significant fraction of the Thr65-containing peptide remained unmodified at the end of the kinase reaction, indicating a higher affinity of PLK1 for Ser46 (Figure 2A and B). To confirm that Ser46 was a phosphorylated site also in cells, a specific antibody against a synthetic peptide containing phospho-Ser46 of TCTP (anti-phospho-TCTP-Ser46) was raised. Initially, the production of an antibody against phospho-Thr65 was also attempted; however, it failed, possibly due to the low antigenicity of the site.

Phosphorylation sites of TCTP resided in different peptides (33-63, 64-80, 59-86). MALDI-TOF analysis of phosphorylated TCTP digested with V8 protease. The ions at m/z 1972.9 (A), 3139.5 and 3157.4 (C) correspond to TCTP phosphorylated peptides 64-80, 59-86 and 33-63 respectively. The ion at m/z 1892.9 (A and B) correspond to the non-phosphorylated form of the peptide 64-80. In B and D, the samples were subjected to phosphatase treatment. Peaks marked with an asterisk were metastable ions detected only in reflector mode. These ions were not detectable after phosphatase treatment and when the instrument was used in linear mode (data not shown).
Anti-phospho-TCTP-Ser46 antibody characterization. The specificity of the anti-phospho-TCTP-Ser46 antibody was assessed by Western blot analysis on recombinant TCTP when phosphorylated by PLK1 and when not. Under these conditions the antibody recognised only the phosphorylated form of TCTP (Figure 3). To eliminate the possibility of a cross-reaction with the secondary phosphorylated site, the specificity was also confirmed on synthetic peptides containing phospho-Ser46, and phospho-Thr65 in a dot-blot experiment (data not shown).
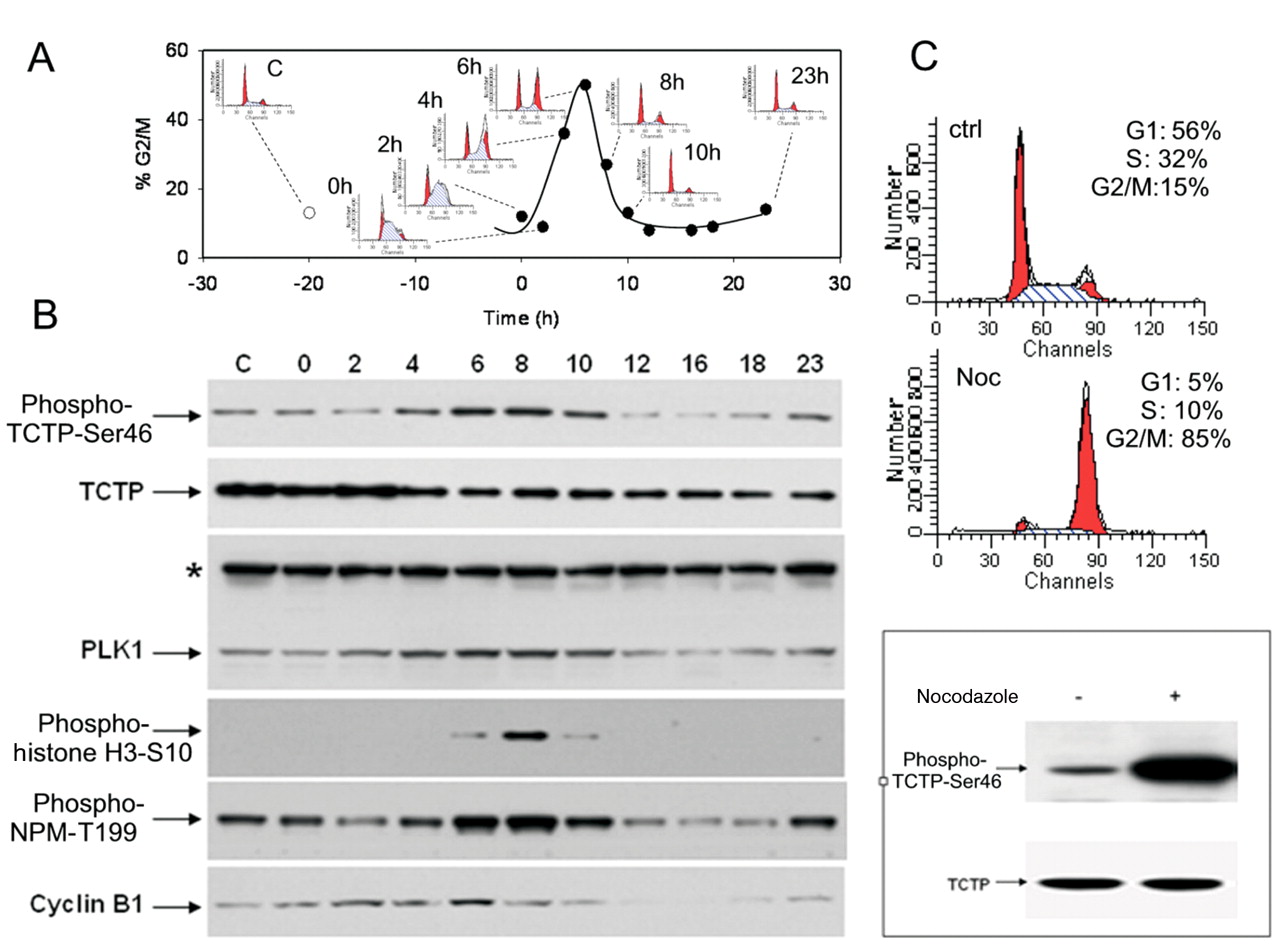
Cell-cycle regulation of TCTP phosphorylation. To verify that TCTP is phosphorylated in cells and this phosphorylation is cell-cycle regulated, the antibody anti-phospho-TCTP-Ser46 was used. A2780 human ovarian adenocarcinoma cells growing asynchronously or synchronised in G1/S phase by thymidine and collected at different time points (0, 2, 4, 6, 8, 10 and 23 h) after release were analysed by flow cytometry in order to evaluate their cell-cycle profile (Figure 4A). In parallel, the same samples were lysed to assess the protein expression by Western blotting (Figure 4B). A clear increase of the phospho-TCTP-Ser46 signal was evident at 6 h (Figure 4B) corresponding to a G2/M accumulation of cells as demonstrated by flow cytometric analysis (Figure 4A). In order to gain insight into the transition from G2 to M phase, specific cell-cycle markers such as cyclin B1, phospho-histone H3-Ser10, phospho-Thr199 nucleophosmin (Phospho-NPM-Thr199) and PLK1 were also analysed. It was found that TCTP phosphorylation started to increase together with cyclin B1 expression at 4 h after thymidine release, correlating with the transition from G2 to M phase. However, while cyclin B1 at 8 h drastically decreased and in parallel phospho-histone H3-Ser10 clearly increased indicating that cells entered mitosis, the phospho-TCTP-Ser46 signal remained high. Even at 10 h after release, when the phospho-histone H3-Ser10 signal returned to its basal level, the phospho-TCTP-Ser46 signal only partially decreased, showing that the phosphorylation is maintained from G2 phase until telophase, covering all mitosis steps as confirmed by phospho-NPM-Thr199 expression (Figure 4B). At the same time points, PLK1 expression increased in a similar manner to phospho-TCTP-Ser46, indicating that PLK1 may be correlated to the phosphorylation of TCTP. The maintained high level of phosphorylated TCTP during mitosis was also confirmed by treatment of cells with nocodazole, a well-known agent, blocking cells in metaphase (Figure 4C).
Biochemical IC50 and cellular potency calculated on phospho-TCTP-Ser46 array scan assay for tested compounds.
In vitro biochemical potency and antiproliferation activity of tested PLK inhibitors.
Phosphorylation of TCTP promotes nuclear localisation. The study investigated whether phosphorylation is able to promote protein relocalisation inside the cell using anti-phospho-TCTP-Ser46 and anti-total TCTP antibodies by immunofluorescence analysis of U2OS cells at different phases of the cell cycle. U2OS human osteosarcoma cells were positive for phospho-TCTP-Ser46 immunostaining from G2 phase (Figure 5C, white arrows, and 5G) to telophase (Figure 5Q), confirming the results obtained by Western blotting. In G2 phase, while total TCTP showed a prevalent cytoplasmic localisation (Figure 5B and F), phospho-TCTP-Ser46 appeared to be mainly nuclear (Figure 5C and G). During mitosis, the immunofluorescence of phospho-TCTP-Ser46 showed a peri-chromosomic localisation (Figure 5M and Q).

Anti-phospho-TCTP-Ser46 antibody was able to detect recombinant TCTP phosphorylated by PLK1. Western blot analysis of non-phosphorylated and phosphorylated TCTP. Phospho-TCTP-Ser46 antibody clearly detected a strong signal only on TCTP phosphorylated by PLK1. The total amount of TCTP was comparable in the presence and in the absence of ATP.
PLK1 depletion or inhibition effects on Ser46 phosphorylation. To evaluate the effect of PLK1 depletion on phospho-TCTP-Ser46 signal, RNA interference (siRNA) experiments were performed. In order to increase the basal level of TCTP phosphorylation, cells were synchronised in mitosis with nocodazole and just a partial reduction of PLK1 protein levels was applied to avoid a strong apoptotic effect, possibly interfering with marker modulation. In fact, accumulation in the G2/M phase but no subG1 induction was observed by flow cytometry analysis in PLK1 siRNA samples in comparison to control samples. Under these conditions, a clear decrease in Phospho-TCTP-Ser46 signal was evident (Figure 6). To confirm this result, PLK1 was inhibited by using the specific PLK1 inhibitor compound 1 (Table II) in synchronised and in asynchronous cells. After 1-h treatment with compound 1 in nocodazole-synchronised cells, a clear reduction of phospho-TCTP-Ser46 signal was observed even though the cells remained blocked in mitosis, as shown by Western blotting with anti-phospho-histone H3-Ser10 and anti-phospho-NPM-Thr199 antibodies (Figure 7A) These data confirmed the direct link between PLK1 and phosphorylation of TCTP. Reduction of phospho-TCTP-Ser46 signal in cells treated with PLK1 inhibitor was not due to protein degradation, as cell treatment with MG 132, a known proteasome inhibitor, did not restore phosphorylation levels detected in the untreated cells (Figure 7A). This observation indicated that PLK1 is the main kinase involved in TCTP phosphorylation on Ser46. The down-modulation of phospho-TCTP-Ser46 was even more evident in asynchronous cells (Figure 7B) and this allowed the use of the phosphorylated protein as a direct marker for evaluating PLK1 activity inhibition other than in cells also in xenograft tumours.
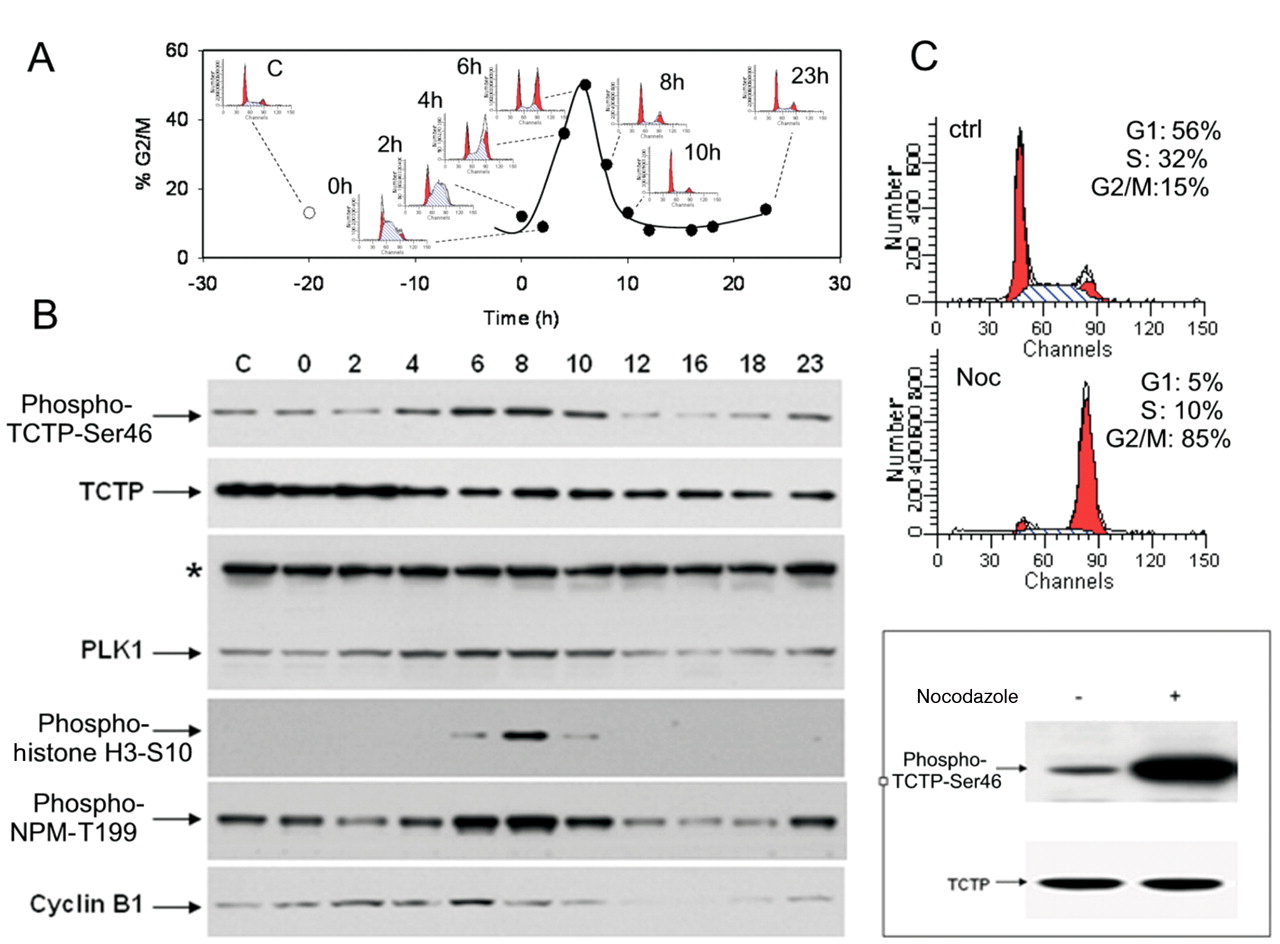
TCTP was phosphorylated on serine 46 during G2 phase and mitosis. A2780 cells were synchronised at the G1/S transition by thymidine treatment. Cells were collected at the indicated time points after release and analysed for DNA content by flow cytometry (A) or lysed for protein expression by Western blotting (B). In A, the percentages of G2/M cells at each time point are reported together with the corresponding cell-cycle profile. In B, protein expression levels for phospho-TCTP-Ser46, total TCTP, total PLK1 and the cell-cycle markers such as phospho-histone H3-Ser10, phospho-NPMThr199 and cyclin B1 are reported. *An aspecific band detected with PLK1 antibody was used as loading control. In C, A2780 cells were synchronised in M phase by nocodazole treatment as demonstrated by cell-cycle profile. Western blot analysis with anti-phospho-TCTP-Ser46 antibody showed a strongly increased expression of phosphorylated protein in cells blocked in mitosis. An antibody against total TCTP was used as a control.
Development of a high content assay for screening PLK1 inhibitors. Since phospho-TCTP-Ser46 is a direct marker of PLK1 activity, the specific antibody was used against this phosphorylated protein to develop an array scan assay to select PLK1 inhibitors in cellular models. The advantage of this kind of assay is that many inhibitors may be analysed in parallel at different doses and an IC50 value for every single compound may be calculated in the specific test. The compounds were first tested on asynchronous cells with a 24-h treatment for phospho-histone H3-Ser10 and cyclin B1 increase to identify molecules that induced accumulation in G2/M phase (Figure 8A). The same compounds were selected for PLK1 inhibition, evaluating phospho-TCTP-Ser46 decrease (Figure 8B) on cells synchronised in mitosis by nocodazole in order to increase the basal level of the phosphorylated protein and then treated for 1 h with the compounds. Comparing the biochemical potency and the cellular activity based on phospho-TCTP-Ser46 assay for PLK1 inhibitors, a good correlation between IC50 was observed. Indeed, a high biochemical potency corresponded to a high activity also in cells, while compounds with biochemical IC50 >100 nM were scarcely active in the array scan assay at the maximal concentration tested of 10 μM (Table I). Using this assay, a large number of molecules were processed and ranked for potency on the specific target in cells making the compound selection easier and faster.

Phosphorylation induced a perichromosomic localisation of TCTP during mitosis. U2OS cells were analysed by immunofluorescence (original magnification, ×40). DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) (A, E, I, O). TCTP and phospho-TCTP-Ser46 were detected with specific antibodies (B, F, L, P and C, G, M, Q respectively). White arrows indicate cells in G2 phase; the red arrow indicates a mitotic cell. E, F, G and H show cells in G2; I, L, M, N represent a cell in metaphase; O, P, Q, R show a telophase cell
Evaluation of phospho-TCTP-Ser46 levels in tumour tissues. In order to evaluate the PLK1 inhibitor activity and their mechanism of action in vivo in preclinical models, the expression levels of phospho-TCTP-Ser46 in xenograft tumours were investigated by immunohistochemistry. Formalin-fixed, paraffin-embedded A2780 tumours were analysed by the specific antibody anti-phospho-TCTP-Ser46 and a clear signal in an evaluable number of cells was observed (Figure 9A) Further analysis for studying the localisation of this phosphoprotein were performed on the same samples by immunofluorescence. Phospho-TCTP-Ser46 was expressed in mitotic cells (Figure 9B) and showed a perichromosomal localisation in the different mitotic phases (Figure 9C, D and E) confirming the finding observed in U2OS cells by immunofluorescence. With regard to the array scan assay, synchronisation of cells in G2/M induced a clear increase of phospho-TCTP-Ser46, in the same way the treatment of A2780 tumour-bearing mice with paclitaxel induced a strong increase of mitotic cells (as revealed by phospho-histone-H3-Ser10 staining), all expressing phospho-TCTP-Ser46 (Figure 10). This result confirmed the validity of this marker also in vivo, paving the way for its future use in the selection of PLK1 inhibitors in preclinical models and in pharmacodynamic studies in clinical trials.
Discussion
In the last 20 years, research for new anticancer agents has been focused on the identification of molecules able to selectively hit specific targets considered to be the drivers for the abnormal proliferation of defined tumour subsets (19). In this new paradigm of cancer research, the availability of specific biomarkers to evaluate the inhibition of the target is of paramount importance. Indeed, in the early stages of drug development, biomarkers are necessary to provide evidence that the reduction of proliferation of cultured cells and the growth of xenografted tumours are directly correlated with the inhibition of the target. After that, a direct biomarker of the target activity will enable pharmacodynamic studies allowing for the identification of the drug dose giving the maximal target inhibition, while avoiding toxicity due to overdosing (20). Recent studies have pointed to the protein kinase PLK1 as an attractive target in anticancer therapy and a set of compounds targeting this protein are now in clinical development.
PLK1 is the master regulator of mitotic progression and inhibition of its activity causes cell-cycle block in mitosis followed by apoptosis. Phosphorylation of histone H3 at serine 10 is a clear hallmark of mitotic cells and has been used to follow the activity of PLK1 in cells and in preclinical in vivo studies performed with BI-2536, BI-6727 and GSK-461364, which are the most clinically advanced PLK inhibitors (21-23). Mitotic block, however, is not a direct marker of PLK1 activity as the same effect is obtained with different antimitotic agents acting on microtubules such as Eg5 inhibitors or even paclitaxel and vinca alkaloids. A more direct marker of PLK1 activity in cells and tissues is still lacking although different substrates are known to be phosphorylated by PLK1 in cells. Indeed, in general, the translation of a bona fide kinase substrate into a preclinically or clinically useful biomarker is not straightforward, being dependent on the availability of specific antibodies or even on the stoichiometry of substrate phosphorylation in vivo.

PLK1 siRNA inhibited TCTP phosphorylation in HCT-116 cells blocked in mitosis. Decreased signal of phospho-TCTP-Ser46 was observed in nocodazole-treated HCT116 cells in which a partial depletion of PLK1 was induced by siRNA. Cell lysates were analysed by Western blotting for TCTP phosphorylation, PLK1 expression and cell cycle markers such as phospho-Histone H3-Ser10, phospho-NPM-Thr199 and Cyclin B1. *An aspecific band detected with PLK1 antibody was used as loading control.
An alternative way to monitor PLK1 inhibition involves measuring its residual activity in cells or tissues following inhibitor treatment by an assay such as the recently described enzyme-linked immunosorbent assay based on the in vitro phosphorylation of the PLK1 substrate peptide PBIP1 (24). However, the availability of a direct marker of PLK1 activity suitable for immunohistochemical studies will help in determining not only the overall level of kinase inhibition but also the specific phenotype and the distribution inside the tissues of the cells in which PLK1 activity is decreased.

TCTP phosphorylation was reduced in cells treated with a PLK1 inhibitor. A2780 cells synchronised with nocodazole (A) or asynchronous (B) were treated with different concentrations of a specific PLK1 inhibitor (compound 1) for 1 hour. Cell lysates were immunoblotted with the indicated antibodies. A clear reduction of phospho-TCTP-Ser46 was observed in both experimental conditions. To verify that the phospho-TCTP-Ser46 inhibition was specifically due to PLK1 inhibitor and not to protein degradation, MG132 was added to the samples of the two last lanes (A). *An aspecific band detected with PLK1 antibody was used as loading control.
In order to find a direct biomarker suitable to follow PLK1 activity in cells and in tumour tissues, the present study was focused on TCTP, a small, highly abundant protein, reported to be an in vitro substrate of PLK1. The aim of the study was first to unambiguously identify the sites phosphorylated in vitro by PLK1 on TCTP by MS, then to raise antibodies against the phosphorylated sites and to validate them in cells and finally to use the generated antibody to follow them as markers of PLK1 activity in cells and possibly in tumours.
The in vitro characterization demonstrated that PLK1 phosphorylates TCTP on two different residues, serine 46 and threonine 65, both consistent with the consensus sequence E/D/Q-X-S*/T*-F described for PLK1 phosphorylation (12). The finding of phosphorylation at threonine 65 is not in agreement with previously published data (16) which indicated serine 64 as a secondary phosphorylated residue in TCTP. This disagreement may have been due to the different approaches used for phosphosite identification. In the present study, direct recognition of phosphorylated sites by MS allowed the unambiguous identification of threonine 65 as the residue phosphorylated in vitro by PLK1, whereas in (16) a silencing point mutation of serine 64 was introduced to demonstrate loss of phosphorylation, which may have had an impact also on the accessibility to phosphorylation of the adjacent residue, possibly by inducing protein conformational changes. This indication reinforced the concept that phosphorylation mapping by point mutation is possible to introduce artifacts in mutant forms due to modification of folding, aggregation status and/or consensus.
The conditions applied for in vitro phosphorylation are usually very different from the intracellular physiological ones due to the presence of sub-cellular compartmentalisation in cells, which may impair interaction between the identified substrate and the target, and the possible engagement of the candidate substrate in protein complexes that may limit the accessibility of the region to phosphorylation. Therefore, the validation in cell of in vitro-identified phosphorylated sites through dedicated antibodies is mandatory. The present study produced and characterised an antibody specific for the main phosphorylated site of TCTP (serine 46). Through this antibody, the signal in cells synchronised in different cell-cycle phases showed that phospho-TCTP-Ser46 and PLK1 expression both peak from G2 to M, demonstrating in this way a correlation between these two proteins. However, a contribution to this effect by other kinases with activity peaking in the same cell-cycle phase may not be ruled out at this point. To exclude this possibility and reinforce the link between the two proteins, experiments of PLK1 silencing were performed. Since more than 90% reduction of PLK1 in tumour cells resulted in G2/M phase cell-cycle block, followed by apoptosis that is usually accompanied by strong non-specific proteolytic activity, a direct effect may be masked by more general protein degradation. For this reason, only a partial silencing of PLK1 was applied. Even when the reduction of PLK1 level was barely appreciable by Western blotting and did not induce an apoptotic effect, it produced a mild but significant decrease of phospho-TCTP-Ser46 signal. This evidence further confirmed that PLK1 is the kinase responsible for TCTP phosphorylation at position 46. An additional validation experiment was run using a specific PLK1 inhibitor and treating the cells for a short time. Also in this case, a significant decrease of phospho-TCTP-Ser46 signal was observed in cells synchronised in mitosis. Moreover, the decreased signal was also clearly observed in asynchronous cells. It was also demonstrated that the decreased phosphorylation is not due to an increase in phosphoprotein degradation, as the treatment with an inhibitor of proteasomal activity did not re-establish phospho-TCTP-Ser46 starting levels.

Array scan assay for screening of PLK1 inhibitors and detecting their potency in cells. A: U2OS cells were treated with PLK1 inhibitors for 24 hours and % of cells positive for phospho-histone H3-Ser10 and cyclin B1 were evaluated. Treatment with potent PLK1 inhibitors (compound 1 and 2) resulted in an increase of both parameters. B: U2OS cells were synchronised in mitosis by an 18-hour treatment with nocodazole then treated for 1 hour with PLK1 inhibitors and evaluated for phospho-TCTP-Ser46 decrease. PLK1 inhibitors produced a dose-dependent decrease in phospho-TCTP-Ser46 signal.
Once the phospho-TCTP-Ser46 antibody was characterized as a tool for revealing PLK1 activity, it was possible to set up a specific high-content assay for screening in parallel different PLK1 inhibitors at different doses, allowing the calculation of the potency of each compound for PLK1 inhibition in cells. Preliminary data obtained with some small-molecule PLK1 inhibitors showed that there is a good correlation between biochemical and cellular potency.
The same antibody was used for immunocytochemical analysis that revealed the presence of phospho-TCTP-Ser46 in cells that morphologically appeared to be in G2 and M phases and showed that its localisation is essentially nuclear and perichromosomal. The same finding was also appreciable in xenograft tumours. Furthermore, the increase of the signal in tumours from mice treated with a therapeutic dose of paclitaxel once again confirmed that TCTP phosphorylation occurs during mitosis. The possibility of using this antibody on formalin-fixed paraffin-embedded tissues will allow the evaluation of the activity of PLK1 inhibitors in future in vivo experiments.
In recent years, the need for evaluating the efficacy and pharmacodynamic behaviour of drugs in clinical development has emerged and, therefore, the availability of a direct marker of PLK1 activity that can be followed not only in cell extracts, but also in preclinical and clinical tumour samples or in surrogate tissues, begins a new phase in PLK1 inhibitor studies In the future, the introduction of phospho-TCTP-Ser 46, a direct pharmacodynamic marker, as a complementary test in combination with phospho-histone H3-Ser10 and with the ELISA assay may help in illustrating the dynamic process of PLK1 activation and deactivation, allowing a comprehensive understanding of the effects of the different extent of PLK1 inhibition.

Phospho-TCTP-Ser46 was detectable in xenograft tumours. Evaluation of phospho-TCTP-Ser46 in A2780 xenograft tumours by immunohistochemistry (brown cells) (A) and by immunofluorescence (red cells) (B). Phospho-TCTP-Ser46 was expressed in mitotic cells with a perichromosomal localisation as detailed in C, D and E. Original magnification, ×400 (A and B).

Phospho-TCTP-Ser46 signal was increased in xenograft tumours from paclitaxel-treated mice. A2780 xenograft tumours from mice treated with vehicle or paclitaxel were stained by immunohistochemistry for phospho-TCTP-Ser46 and phospho-histone H3-Ser10. Paclitaxel induced a strong increase of mitotic cells, as demonstrated by phospho-histone H3-S10 staining (compare A and B), which all expressed phospho-TCTP-Ser46 (D). Original magnification, ×200. Insets show mitotic cells at higher magnification.
Acknowledgements
We thank Rosario Baldi and Silvia Messali for technical assistance, Roberta Bosotti for bioinformatic analysis, Antonella Isacchi and Francesco Colotta for support, Vanessa Marchesi, Luisa Rusconi and Clara Albanese for critical reading of the manuscript.
Footnotes
-
↵* Current address: IFOM-IEO Campus, 20139, Milan, Italy.
- Received September 30, 2010.
- Revision received November 5, 2010.
- Accepted November 8, 2010.
- Copyright© 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Chk2 sustains PLK1 activity in mitosis to ensure proper chromosome segregation
- A Phase Ib Study of Onvansertib, a Novel Oral PLK1 Inhibitor, in Combination Therapy for Patients with Relapsed or Refractory Acute Myeloid Leukemia
- DNA replication and mitotic entry: A brake model for cell cycle progression
- Efficacy and Mechanism of Action of Volasertib, a Potent and Selective Inhibitor of Polo-Like Kinases, in Preclinical Models of Acute Myeloid Leukemia
- Personalizing the Treatment of Pediatric Medulloblastoma: Polo-like Kinase 1 as a Molecular Target in High-Risk Children
- NMS-P937, an Orally Available, Specific Small-Molecule Polo-like Kinase 1 Inhibitor with Antitumor Activity in Solid and Hematologic Malignancies
- Polo-like Kinase 1 Inhibitors and Their Potential Role in Anticancer Therapy, with a Focus on NSCLC