


Abstract
Motility of endothelial cells is a requirement for the vascularization of solid malignancies. While tumors have been shown to produce a host of angiogenic factors, including TGF-β, the mechanisms by which such factors regulate endothelial cell motility have not yet been defined. Thus, the role of the serine/threonine phosphatase PP-1 in regulating endothelial cell motility and cytoskeletal architecture was studied. The present study demonstrated that TGF-β stimulation of motility is dependent on PP-1. Likewise, TGF-β was shown to up-regulate paxillin expression through a process that was PP-1 dependent. The interplay between PP-1 and TGF-β was further observed by the induction of cell rounding and the loss of paxillin-actin co precipitations upon PP-1 inhibition and the compensation for these effects by TGF-β. Studies initiated to determine how PP-1 might regulate motility showed its role in maintaining cytoskeletal organization and its capacity to directly dephosphorylate the focal adhesion scaffolding protein paxillin. These studies suggest that the interplay between TGF-β and PP-1 regulates the motility of endothelial cells that is critical to the process of angiogenesis.
Motility of endothelial cells is a critical component of the process of tumor-induced angiogenesis. Treatment of endothelial cells with tumor-conditioned media has previously been shown to stimulate their motility (1). TGF-β is a significant component of the tumor-conditioned medium responsible for stimulating endothelial cell motility, since tumor-conditioned media that are depleted of TGF-β have a more limited capacity to induce microvascular endothelial cell migration (1). TGF-β has been shown to stimulate the motility of not just endothelial cells, but also a variety of other cell types such as keratinocytes, fibroblasts and cancer cells (2, 3, 10, 11). Focal adhesions are critical signaling complexes for cellular migration. The function of the focal adhesion complex relies on the coordinated participation of a multitude of proteins such as FAK, paxillin, p130Cas, Src, and many others (4). Paxillin, however, has been of interest as it is a crucial focal adhesion scaffold, coordinating with signaling and cytoskeletal networks.
Paxillin has been shown to be multiply phosphorylated, leading to its ability to interact with a diverse host of targets (9). Its phosphorylation occurs on tyrosine, serine and threonine residues, although tyrosine phosphorylation has been more extensively studied as it appears to be important in coordination of paxillin with focal adhesion kinase and other kinase and second messenger proteins with functional consequences on cellular cytoskeleton and adhesiveness (14). The function of serine and threonine phosphorylation has also been shown to be important as it is involved in localization of paxillin to the focal adhesion (6). The function consequence of changes in paxillin phosphorylation and alterations in the focal adhesion architecture is that cellular migration is a balance between stabilized focal adhesions to anchor and focal adhesion turnover to migrate.
Treatment of endothelial cells with tumor-conditioned media has been shown to reduce the activity of the serine/threonine phosphatase PP 2A, which is sufficient to stimulate cellular motility (13). PP 2A has previously been shown to be important in paxillin dephosphorylation (7). However, whether the serine/threonine phosphatase PP-1 regulates cellular motility is less well understood. While TGF-β signaling can increase PP-1 activation, the implications of such findings on cellular migration have not been addressed (5).
The present study showed that PP-1 was able to dephosphorylate paxillin. While TGF-β stimulated the motility of endothelial cells, this stimulated migration was dependent on PP-1. The interplay between PP-1 and TGF-β was also observed by TGF-β blocking PP-1-mediated alterations in cellular morphology and localization of paxillin with actin. These studies suggested the contribution of the interplay between TGF-β and PP-1 in regulating the motility of endothelial cells.

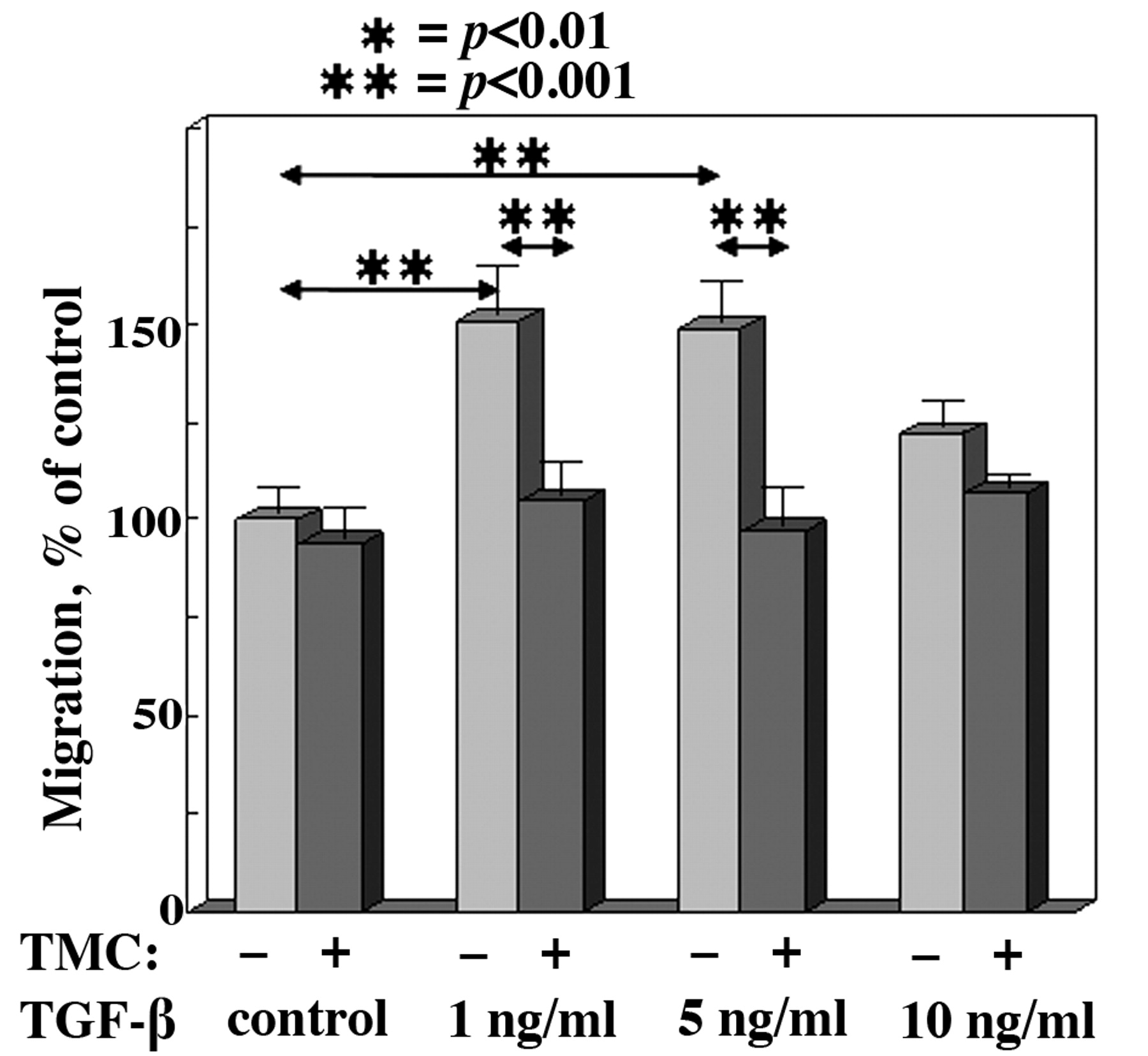
TGF-β–mediated endothelial cell migration is PP-1 dependent. The migration of LLC cells from the upper compartment to the lower compartment of a transwell migration chamber was assessed in the presence of various doses of TGF-β with or without 500 nM tautomycetin (TMC) to inhibit PP-1 activity. Migration is expressed as mean percentage, normalized to the control-treated endothelial cells. Error bars represent SEM for triplicate measurements in 3 separate experiments.
Materials and Methods
Cells and media. The murine microvascular brain endothelial cell line, bEnd.3 (ATCC, Manassas, VA, USA), was grown in DMEM culture medium (Invitrogen, Carlsbad, VA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, 0.02 M HEPES buffer, 2 mM L glutamine, and 5×10−5 M 2-mercaptoethanol. A 0.05% trypsin, 0.53 mM EDTA solution (Invitrogen) was used to detach endothelial cells from the culture flasks prior to passage. For migration assays, Accutase (MP Biomedicals, Solon, OH, USA) was instead used to detach cells from culture flasks prior to plating in transwell plates.
A metastatic clone of Lewis lung carcinoma (LLC) was grown in RPMI culture medium (Invitrogen) supplemented identically to the medium used for the endothelial cell line. Tumor conditioned media was generated by collecting the supernatants from 24 hr cultures of LLC cells.
Treatments. Prior to use, endothelial cells were cultured for 24 h in reduced-serum DMEM medium containing 0.5% FBS. Endothelial cells were then treated with recombinant human TGF-β1 (R&D Systems, Minneapolis, MN, USA) and/or with 500 nM tautomycetin (Tocris, Ellisville, MO, USA) as a selective PP-1 inhibitor. Medium containing DMSO was used as the diluent control for tautomycetin.
Enzyme-linked immunosorbent assays (ELISA). TGF-β1 secreted by LLC tumor cells into culture medium and the levels of TGF-β1 in LLC lysates was measured using a TGF-β1 BD OptEIA assay kit (BD Bioscience, San Diego, CA) according to the manufacturer's instructions. Samples were acid activated prior to measuring TGF-β levels.
Transwell migration assay. Endothelial cells that were incubated in serum-reduced medium for 24 h were detached with Accutase and plated at a density of 5×104 cells into the top compartment of a transwell migration chamber. Both the upper and lower wells contained diluent or TGF-β and/or 500 nM tautomycetin in reduced-serum DMEM. After overnight migration, endothelial cells were collected from the lower compartment of the chamber and the relative number of cells was determined using the CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI, USA). Relative optical density of the colored product correlates to the number of viable cells. Results were reported as the percent of the value for control cells.
Phosphatase assay. PP-1 activity was measured using the ProFluor serine/threonine phosphatase assay kit (Promega) containing appropriate salts selective for PP-1 activity. Purified PP-1 (Millipore, Billerica, MA, USA) was used for the standard curve. Endothelial cell lysates were generated in RIPA buffer on ice. Lysates were measured for protein content and equal quantities of protein were diluted in the provided dilution buffer containing appropriate salts for PP-1. A selective inhibitor of PP 2A was added to lysates to eliminate non-specific activity. To each sample and standard, a fluorescently labeled substrate was added for 10 min. The reaction was stopped using the supplied protease buffer, releasing the fluorescent tag from dephosphorylated substrates. Fluorescence was measured on a SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, CA, USA). Phosphatase activity was calculated as mU/μl. In this assay 1 U is equivalent to 1nM phosphate released per minute.
Immunoblotting. Endothelial cells were rinsed with cold PBS and removed by scraping in the presence of RIPA buffer containing 1X PhosphoStop and MiniComplete phosphatase and protease inhibitor cocktails (Roche, Basel, Switzerland). Lysates were vortexed and incubated for ten minutes on ice. Cell debris was pelleted by centrifugation at 14,000 rpm for 30 min at 4°C. Protein content of the lysates was measured using a BCA protein assay (Pierce, Rockford, IL, USA) to equalize protein content of loaded samples to 25 μg for whole cell lysates. For lysates being used for immunoprecipitation, 250 μg of sample was mixed with 50 μl magnetically labeled Protein G microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) and 2 μg of mouse anti paxillin antibody (BD Transduction Labs). The mixture was incubated for 30 min on ice. The microbeads were placed on a magnetic separation column and flow through fractions were collected. The beads were washed four times with RIPA buffer and twice with 25 mM TRIS buffer, and then eluted for separation by SDS PAGE in hot Laemelli sample buffer. Protein bands were resolved on 10% or 7.5% polyacrylamide gel after passing through a 3% stacking gel. Proteins were transferred to nitrocellulose membranes for Western blotting. Membranes were blocked for 30 min at 37oC in a 5% BSA in TRIS buffered saline with 0.5% Tween-20 (TBST). Primary antibodies were added to the blocking solution and incubated overnight at 4°C. Membranes were washed three times for ten minutes in TBST and then incubated for one hour at room temperature with the appropriate horseradish peroxidase (HRP) tagged secondary antibody dissolved in a 5% solution of BSA in TBST. An enhanced chemiluminescence (ECL) reagent (GE) was used to detect antibody tagged proteins using the VersaDoc Imaging System (Bio Rad, Hercules, CA, USA). Antibodies used in these studies were raised against β actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:1,000), phosphoserine (Calbiochem, San Diego, CA, USA; 1:500), paxillin (BD Transduction Labs 1:2,000), pan-specific PP-1c (Santa Cruz Biotechnology 1:200), HRP-goat anti-mouse IgG (Millipore, 1:1,000), and HRP-goat anti mouse IgM (Invitrogen, 1:1,000).
Paxillin dephosphorylation. Endothelial cells were incubated in reduced-serum medium for 12 h and then treated with 100 nM okadaic acid for 10 h to inhibit PP 2A activity. An additional two hour incubation was performed with 100 nM okadaic acid, 5 μM tautomycetin, and 50% LLC conditioned media to maximize serine and threonine phosphorylation. Cells were rinsed with cold PBS and lysed in RIPA buffer. Diluent or purified PP-1 (0.5 Units, Millipore) was added to 25 μg aliquots of whole cell lysates or paxillin immunoprecipitated from 250 μg lysates and suspended in PP-1 dilution buffer (Millipore). After incubation at 37°C for 30 min to allow for dephosphorylation, immunoblotting was carried out as described above.
Results
TGF-β stimulation of endothelial cell migration. Media conditioned by murine LLC cells has previously been shown to increase endothelial cell motility. Neutralization of TGF-β in the conditioned media diminishes this stimulation of motility (13). Therefore, levels of TGF-β produced by a metastatic LLC clone were measured. While control medium contains TGF-β due to the presence of 10% FBS, LLC cells secreted 652+95 pg/ml more TGF-β than was present in medium alone. Further, LLC lysates contained 224+13 pg/ml TGF-β. Because of the TGF-β content in FBS, subsequent studies pre incubated endothelial cells in serum-reduced medium (0.5%) before their use.
Since TGF-β can stimulate endothelial cell motility (13) and since serine/threonine phosphatases have previously been shown to regulate cellular motility (12), the possibility of an inter-relationship between TGF-β and the phosphatase PP-1 was examined. The contribution of PP-1 activity to TGF-β-induced migration was determined by using tautomycetin to selectively inhibit PP-1. Tautomycetin has previously been shown to exert little to no inhibition of PP-2A activity at doses that completely inhibit PP-1 activity (8). TGF-β treatment at 1 ng/ml and 5 ng/ml increased endothelial migration as compared to control treatment (Figure 1). However, the extent of stimulation by 10 ng/ml TGF-β was not significant. Tautomycetin alone had no statistically significant effect on basal levels of endothelial cell migration. Of significance was the complete abolishment of TGF-β-induced migration by tautomycetin, with migration being statistically the same as for control cells (p>0.05 versus control).

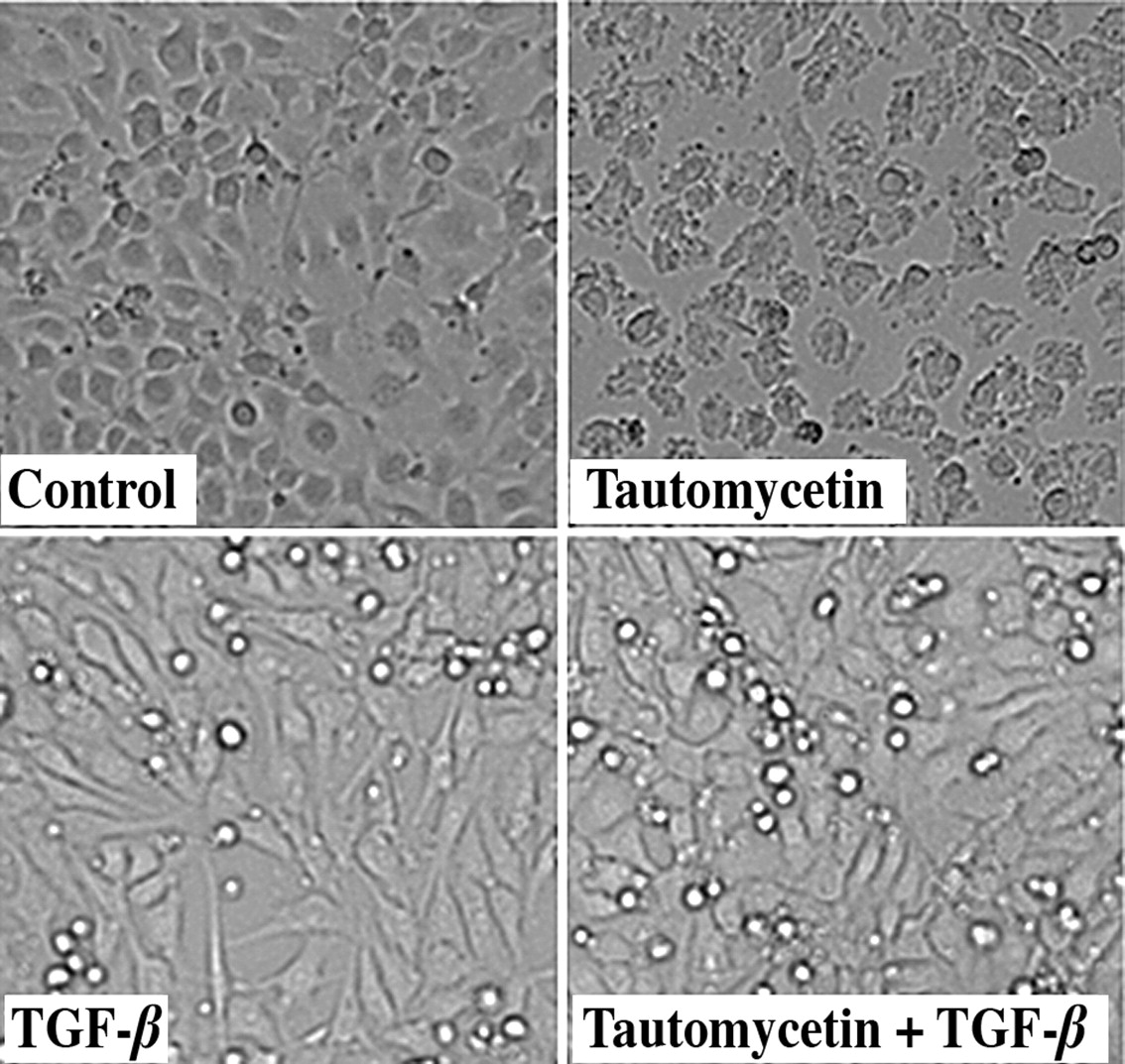
TGF-β prevents tautomycetin-induced endothelial cell rounding. The morphology of endothelial cells that were treated for 24 hours with 10 ng/ml TGF-β and/or 500 nM tautomycetin is shown.
Whether or not TGF-β-stimulated motility might be due to its regulation of PP-1 enzyme activity was examined. Endothelial cells that were treated with TGF-β had a slight reduction in PP-1 activity, although it did not appear to be dose dependent. Specifically, treatment with 1, 5, or 10 ng/ml TGF-β reduced endothelial cell PP-1 activity respectively by 21.7+1.2% (p<0.05), 13.0+1.3% (not significant), and 17.4+0.9% (p<0.05). Thus, the dependence of TGF-β-stimulated motility on PP-1 was not due to a stimulation of PP-1 activity by TGF-β.
Reversal of tautomycetin-induced cell rounding by TGF-β. Since TGF-β stimulation of endothelial cell motility is prevented by PP-1 inhibition, the inter-relationship between TGF-β and PP-1 was explored further by determining the effect of TGF-β on cellular morphology. In contrast to expectation that the increased motility of TGF-β-treated cells would be accompanied by reduced cell spreading, endothelial cells that were treated with TGF-β retained a relatively normal appearance with extensive cell spreading (Figure 2). However, endothelial cells treated with tautomycetin acquired a rounded morphology. This was not the result of toxicity since the cells remained viable and the cells rapidly reverted to a spread morphology following removal of the tautomycetin. Of interest was that adding TGF-β prevented the cellular rounding that occurred by treatment with tautomycetin alone, suggesting a compensatory effect of TGF-β for the inhibition of PP-1 activity.

Representative blots of paxillin Western blot analyses. Top panel: TGF-β increases paxillin protein levels and proportionally increases paxillin co precipitation with actin. Endothelial cells were treated with TGF-β for 24 hours. Paxillin immunoprecipitates (IP) and flow-through fractions from these treated cells were separated and blotted with anti-paxillin, anti-actin, and anti-pan PP1c antibodies. Middle panel: Tautomycetin blocks the TGF-β-induced increase in paxillin levels and prevents the loss of paxillin-actin co-localization that is caused by TGF-β. Endothelial cells were treated with 10 ng/ml TGF-β and/or 500 nM tautomycetin. Whole cell lysates and paxillin immunoprecipitates were blotted with anti-paxillin and anti-actin antibodies. Bottom panel: PP-1 dephosphorylates paxillin. Lysates were exposed to purified PP-1. Whole cell lysates or paxillin immunoprecipitates (IP) and the supernatant (Sup) were blotted for phosphoserine and paxillin.
TGF-β treatment up-regulates paxillin expression, but has no effect on PP-1 expression in endothelial cells. The above results that showed an interplay between TGF-β and PP-1 in regulating endothelial cell motility and morphology, and prior studies that showed that paxillin phosphorylation impacts on focal adhesion architecture and, in turn, motility, prompted assessment of the effect of TGF-β on levels of paxillin and its association with either PP-1 or the actin cytoskeleton. After treating endothelial cells with either diluent or 1, 5, or 10 ng/ml TGF-β, cells were lysed and either whole cell lysates or paxillin immunoprecipitates were blotted for paxillin, actin and PP-1. What was unexpected was an increase in total paxillin levels in TGF-β-treated endothelial cells (Figure 3, top panel). In contrast, TGF-β treatment had no effect on protein levels of PP-1 or actin in whole cell lysates. Results of paxillin immunoprecipitates did not reveal stable co-localization of PP-1 with paxillin. However, there was actin co-precipitation with paxillin. While the levels of actin that co-precipitated with paxillin increased in TGF-β-treated cells, this was proportional to the increased levels of paxillin.
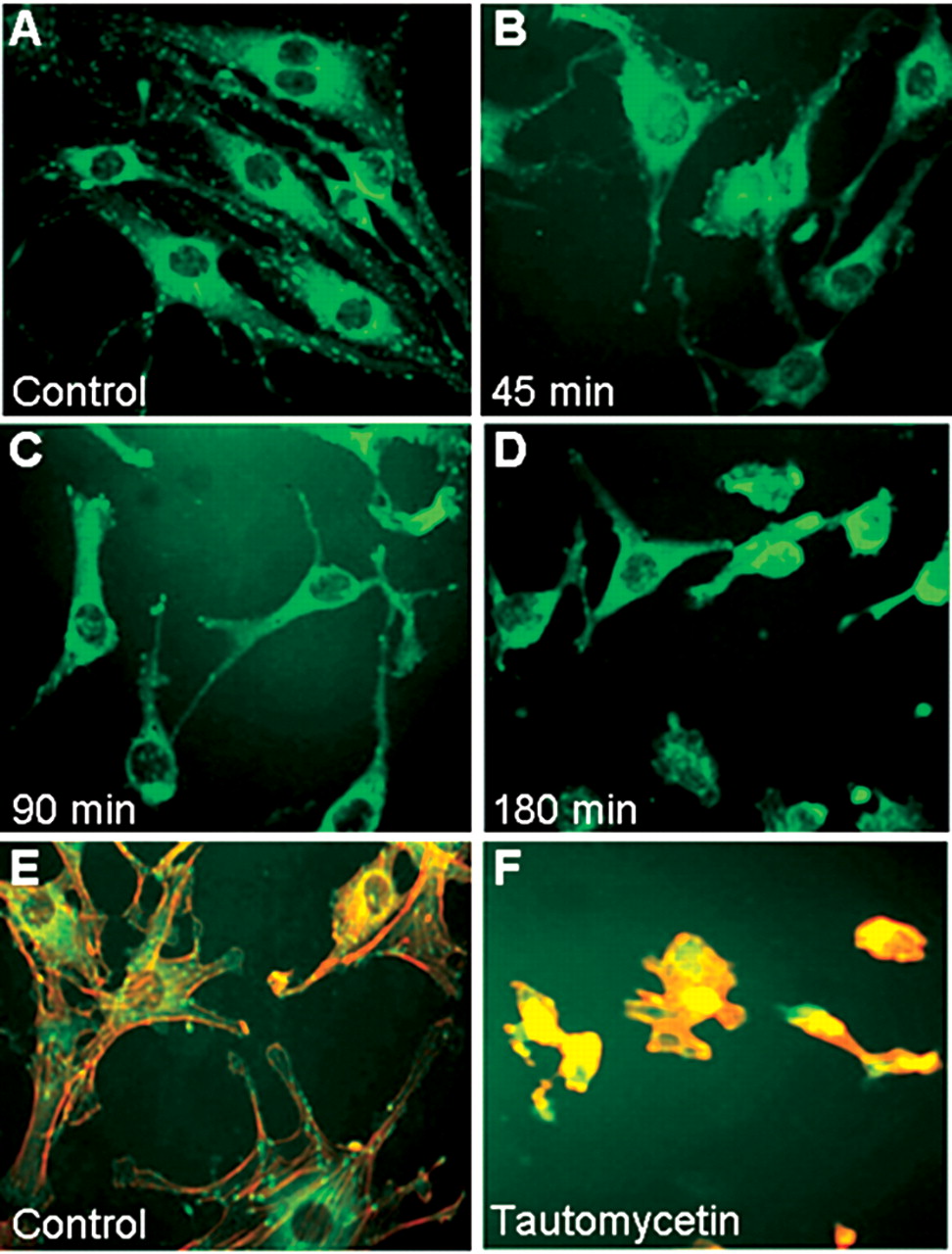
Tautomycetin alters the cytoskeletal organization of endothelial cells. A D. Endothelial cells were treated with 500 nM tautomycetin for the stated times. Paxillin (green) immunostaining was captured by fluorescent microscopy. E F. After treating endothelial cells for 24 h with DMSO diluent control or with 500 nM tautomycetin, they were fixed and immunostained for actin (red) or paxillin (green).
PP-1 inhibition blocks TGF-β-mediated upregulation of paxillin expression and reduces co localization of paxillin with actin. The above studies showing the interplay between TGF-β and PP-1 in regulating cellular motility and morphology prompted determination of whether TGF-β-mediated upregulation of paxillin levels and the association of paxillin with actin were also modulated by PP-1. TGF-β treatment of endothelial cells increased paxillin protein levels (Figure 3, middle panel), as was shown in the top panel (Figure 3). Treatment with tautomycetin alone did not alter paxillin expression. Of significance is that the TGF-β-stimulated upregulation of paxillin levels was dependent on PP-1 activity since treatment of endothelial cells with both TGF-β and tautomycetin blocked the TGF-β-mediated up-regulation of paxillin levels.
Since the above studies showed PP-1 and TGF-β regulate endothelial cell paxillin expression, their regulation of paxillin and actin co localization was also examined. While treatment of endothelial cells with TGF-β had no effect on the co precipitation of actin with paxillin, treatment with tautomycetin abolished the paxillin-actin co precipitation (Figure 3, middle panel). Treating endothelial cells with both tautomycetin and TGF-β, prevented the tautomycetin-induced loss of paxillin-actin co precipitation. These results are consistent with the above demonstration of endothelial cell rounding when PP-1 activity was inhibited and how TGF-β compensates for the loss of PP-1 blockage to restore a spread morphology.
Tautomycetin disrupts paxillin localization to the focal adhesions. The mechanisms by which PP-1 might regulate cellular motility and morphology are largely unexplored. Therefore, studies turned to focusing on the role of PP-1 in the regulation of focal adhesions and the cytoskeleton. Since paxillin phosphorylation is important in cellular adhesion and migration (9), studies first assessed if PP-1 modulates paxillin cellular localization. Endothelial cells were treated with tautomycetin and then immunofluorescent stained to visualize paxillin (Figure 4, panels A-D). After as little as 45 minutes of treatment with tautomycetin, the appearance of focal adhesions became diffuse and paxillin was relocating to the cytoplasm. At 90 minutes, the effect of focal adhesion loss was more evident as a rounded cellular morphology began to appear. Finally, at 180 minutes, focal adhesions were all but lost with many cells rounded and some detached. This loss of paxillin localization and the rounding of tautomycetin-treated cells was not the result of cell death since removal of the tautomycetin restored the spread morphology typical of control endothelial cells.
The effect of PP-1 inhibition on the actin cytoskeletal architecture was also examined. Double staining for actin and paxillin in diluent-treated endothelial cells showed a distinct actin cytoskeleton that associates with paxillin staining within focal adhesions (Figure 4, panels E-F). However, within 180 minutes of treatment with tautomycetin, cytoplasmic extensions and ruffles became contracted and cells became rounded. These studies suggest that the decline in serine/threonine dephosphorylation leads to dissociation of the paxillin-actin localization.
Paxillin is a target of PP-1-directed dephosphorylation. Since the above studies showed an interplay between PP-1 activity, paxillin, and the cellular architecture, studies assessed if paxillin was a phosphorylation target of PP-1. Lysates of endothelial cells were analyzed for the level of paxillin serine phosphorylation. Phosphorylation in the 65-68 kD region, where paxillin localizes, was prominent in lysates of control endothelial cells (Figure 3, bottom panel). However, there is a dramatic decrease in paxillin phosphorylation as a result of adding enzymatically active PP-1 to the endothelial cell lysates.
To confirm the results of the above immunoblots, endothelial cell lysates were treated with or without PP-1, paxillin was immunoprecipitated and blotted for phosphoserine. Similar to the results with whole cell lysates, addition of PP-1 resulted in decreased phosphoserine levels of immunoprecipitated paxillin (Figure 3, bottom panel). These results suggest paxillin to be a target of PP-1-mediated dephosphorylation.
Discussion
This study aimed to determine the contribution of the serine/threonine phosphatase PP-1 in the regulation of the cytoskeletal architecture and on motility of endothelial cells. The rationale for this study was that motility, which is a required component of endothelial cell responses in angiogenesis, is dependent on cytoskeletal rearrangements. While tumor-secreted growth factors are known to influence various cellular signaling networks, signal transduction through protein kinases has been far more extensively studied than the roles of protein phosphatases.
Prior and current studies show that LLC tumors, like other tumor types, produce TGF-β, which stimulates the motility of endothelial (1). Because control FBS-containing medium contains TGF-β, studies were conducted with endothelial cells that were pretreated with serum-reduced (0.5%) culture medium. Previously, it wasshown that both tumor-conditioned medium as well as TGF-β inhibit the activity of the phosphatase PP-2A, which results in stimulated cellular (1, 13). This raised the possibility that PP-1, which is also a serine/threonine phosphatase, may likewise be involved in regulating motility. While the present study did not demonstrate a significant effect of PP-1 inhibition on endothelial cell motility, it did demonstrate an interesting inter-relationship between PP-1 and TGF-β. What was unexpected was that the TGF-β-stimulated motility of endothelial cells was dependent on PP-1 activity. Also unexpected was the upregulation of paxillin expression by TGF-β and the dependence of this upregulation on PP-1.
Paxillin is a scaffolding protein within focal adhesions for a multitude of signaling mediators. Previous studies showed the importance of paxillin phosphorylation on localization to the focal adhesions. However, the role of PP-1 in paxillin phosphorylation or its function as a scaffolding protein has not been studied extensively. The current studies demonstrate that the addition of purified PP-1 to endothelial cell lysates reduces the levels of paxillin serine phosphorylation. However, it was not possible to attribute dephosphorylation of paxillin by PP-1 within the cell to stable binding of PP-1 with paxillin as they did not co precipitate. Instead, dephosphorylation might be occurring through transient association of PP-1 with paxillin or stable association with a spatially proximal protein. When endothelial cells were treated to inhibit PP-1 activity, intracellular paxillin localization was dramatically altered. Specifically, there was a redistribution of paxillin from the focal adhesions and a contraction of actin filaments, leading to endothelial cell rounding. Cellular rounding can be associated with a migratory phenotype (1). However, the migration of tautomycetin-treated endothelial cells was the same as that for untreated cells. A potential reason for rounding of tautomycetin-treated cells without migration may lie in the extent of cell rounding. If rounding is sufficiently extensive to result in cell detachment, then the capacity of cells to form new adhesion sites at the leading edge while detaching at the trailing edge to allow forward motion cannot occur. One explanation of the paradoxical compensation of tautomycetin-induced cellular rounding of tautomycetin-treated cells by the addition of TGF-β may lie in the multitude of signaling pathways initiated by TGF-β. While tautomycetin selectively inhibits PP-1, TGF-β signaling is more complex. The highly regulated cascade of pathways initiated in response to TGF-β might compensate for the inhibition of PP-1, thus preventing the cells rounding that occurs upon PP-1 inhibition.
Among the consequences of inhibiting endothelial cell PP-1 activity was inhibition of actin co immunoprecipitation with paxillin. Similar to its effect on cell morphology, TGF-β treatment compensated for the PP-1 inhibition and prevented the loss of actin co immunoprecipitation with paxillin. Yet to be defined is the network of signaling mediators by which TGF-β neutralizes the effects of PP-1 inhibition to stabilize the actin cytoskeleton and maintain cell morphology. As focal adhesions are crucial for cellular morphology and motility, understanding the role PP-1 on focal adhesions may be important in limiting endothelial cell motility in angiogenesis.
In conclusion, the present study showed a dependence on the serine/threonine phosphatase PP-1 for TGF-β stimulation of endothelial cell motility and upregulation of the focal adhesion scaffolding protein paxillin. In contrast, while PP-1 inhibition resulted in cell rounding and a loss of actin co localization with paxillin, TGF-β compensated for these effects of PP-1 inhibition to prevent cell rounding and the loss of actin-paxillin co localization. Not yet defined is the network of signaling mediators by which TGF-β compensates for the effects of PP-1 inhibition. This newly demonstrated inter-relationship between PP-1 and TGF-β contributes to the understanding of mechanisms that mediate endothelial cell motility in the context of tumor-induced angiogenesis and identifies additional targets for inhibiting angiogenesis and tumor progression.
Acknowledgements
This study is part of the doctoral dissertation of Jarrett Walsh. The study was supported by the Clinical Science and Biomedical Laboratory Research and Development Services of the Department of Veterans Affairs and by grants R01 DE018168 and R01CA128837 from the National Institutes of Health to MRIY.
- Received October 1, 2010.
- Revision received November 1, 2010.
- Accepted November 2, 2010.
- Copyright© 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}